





Abstract
The PRINTS database, now in its 21st year, houses a collection of diagnostic protein family ‘fingerprints’. Fingerprints are groups of conserved motifs, evident in multiple sequence alignments, whose unique inter-relationships provide distinctive signatures for particular protein families and structural/functional domains. As such, they may be used to assign uncharacterized sequences to known families, and hence to infer tentative functional, structural and/or evolutionary relationships. The February 2012 release (version 42.0) includes 2156 fingerprints, encoding 12 444 individual motifs, covering a range of globular and membrane proteins, modular polypeptides and so on. Here, we report the current status of the database, and introduce a number of recent developments that help both to render a variety of our annotation and analysis tools easier to use and to make them more widely available.
Database URL:www.bioinf.manchester.ac.uk/dbbrowser/PRINTS/
Introduction
The PRINTS database has had a long history. The first collection of protein family fingerprints was released in October 1991 as the Features Database, part of the SERPENT information storage and analysis resource for protein sequences established at the University of Leeds (1). At that time, the database contained 29 entries; two-thirds of these were linked to equivalent entries in PROSITE (2), which then held 441 family descriptions. Although disparate in size, the Features and PROSITE databases had various aspects in common; most notable was the principle of added-value through hand-crafted annotation of their diagnostic signatures.
In 1992, motivated by common ideals, and faced with relentlessly time-consuming manual-annotation tasks, we devised a plan to create an integrated protein family resource by merging PRINTS and PROSITE. This vision was finally realized in 1999 with a beta release of a unified database containing 2423 entries—this was InterPro (3). By that time, PROSITE and the Features Database had both undergone significant changes: PROSITE had seen 3-fold growth to 1370 entries (release 16.0); meanwhile, the Features Database had grown 40-fold to 1157 entries (release 23.1) and had been renamed ‘PRINTS’ (4). Therefore, the first release of InterPro combined the contents of PROSITE 16.0 and PRINTS 23.1; in addition, it incorporated family descriptors from 241 profiles, together with 1465 hidden Markov models from Pfam 4.0 (5).
The availability of such diverse resources offers users a range of diagnostic opportunities, from single functional sites and motifs, to complete domains and autonomous folding units, through to extensive domain- and gene-family hierarchies. Inevitably, these different approaches have particular strengths and weaknesses. Fingerprints, for example, by virtue of containing multiple motifs, derive much of their potency from the context afforded by matching motif neighbours, making them generally more flexible and powerful than single-motif approaches. Although they cannot compete with profile-based methods in terms of sensitivity for diagnosing protein superfamilies, they are inherently well-suited to the creation of ‘hierarchical’ discriminators: the fingerprint technique can readily be used to focus on small, uniquely conserved regions that differ between highly similar homologues within closely related families – for example, this approach has been used to resolve G-protein-coupled receptor (GPCR) superfamilies into their constituent families and receptor subtypes (6), and to subclassify and functionally characterize a variety of structural proteins (7), transporters and channel proteins (8), and enzymes (9, 10). Family hierarchies such as these form the backbone of the PRINTS database, and help to structure the organization of gene families within InterPro; in turn, these are used by UniProt to provide automatic annotation for UniProtKB/TrEMBL.
PRINTS was originally built as a single ASCII (text) file. To facilitate maintenance, we later developed a relational version of the resource, known as PRINTS-S (11); in an attempt to increase its coverage, we also created an automatic supplement, termed prePRINTS (12). The main search tools we made available were: a BLAST (13) server, for searches against sequences matched in the current version of the database (14); and the FingerPRINTScan suite (15), for searches against fingerprints contained in the current release (this affords greater specificity than the BLAST implementation). A particularly powerful aspect of FingerPRINTScan is to make explicit the familial hierarchies encoded in PRINTS-S, allowing associations to be traced from subfamily, through family, to superfamily relations and, where relevant, to putative distantly related clan members that share no significant sequence similarity (16). Versions of PRINTS are also still available for searching in Blocks format at the Fred Hutchinson Cancer Research Center (17), via the eMOTIF3.6 and eMATRIX2.0 search tools at Stanford (18), and via InterPro (19), to which it provides a significant amount of annotation and much of its hierarchical information.
Many of these developments, and the significant expansion of PRINTS between 1993 and 2002, were made possible by buoyant funding, and adherence to a strict regime of quarterly releases, adding 200 new entries per year; supporting this rate of growth, however, was not always feasible (in some subsequent years no releases were possible). Now entering its 21st year, PRINTS has moved on and continues both to contribute unique functionality to InterPro and to offer a range of new analysis and curator-assistant tools. Here, we review the main developments of PRINTS and its supporting analysis and annotation software since 2003.
Database developments
PRINTS is released in major and minor versions: minor releases reflect updates, either incorporating a range of trivial typographical and/or format corrections, or more significant changes, such as bringing the entire contents in line with the current version of the source database [a UniProtKB/Swiss-Prot-TrEMBL composite (20)]; major releases denote the addition of new material to the resource, each including around 50 new annotated families (and usually upgrades of fingerprints whose performance has eroded over time). Six major and two minor releases have been made since the last published report, adding 356 new fingerprints (encoding 1513 motifs) and 74 updated fingerprints to the resource.
The evolution of PRINTS has been particularly helped in recent years by productive collaborations with a variety of other projects and databases. Most recently, within the context of the European IMPACT project, PRINTS became more tightly integrated with InterPro: as well as providing new entries to the resource, a novel hierarchy-based filtering approach was developed, helping PRINTS meet InterPro’s automated sequence analysis requirements by resolving protein family membership as unambiguously as possible. As part of the European Kidney and Urine Proteomics project (EuroKUP), we explored a range of medically relevant protein families: aiming to gain a better understanding of specific sequence attributes that may contribute to renal abnormalities or that underpin chronic kidney disease, we developed hierarchical fingerprints for families such as notch, aquaporin and RNA (C5-cytosine) methyltransferases, all of which play active roles in disease-related pathways.
A simple example of how such hierarchies may add value to UniProtKB functional diagnoses (which are derived automatically from InterPro and its source databases) is illustrated in Figure 1. The UniProtKB/TrEMBL entry, Q9NSV5_HUMAN, was annotated as a putative uncharacterized protein; the family and domain database cross-references pointed to its membership of the major intrinsic protein (MIP) superfamily (which contains more than 7000 members), but provided no family-specific information. Viewed with FingerPRINTScan, however, the sequence is quickly diagnosed not only as a member of the MIP superfamily, but specifically also as an aquaporin 6 subtype. This diagnosis was recently supported with an update of InterPro, which included a new version of the PANTHER database (21), whose entry PTHR19139:SF36 also points to membership of the aquaporin 6 subfamily.
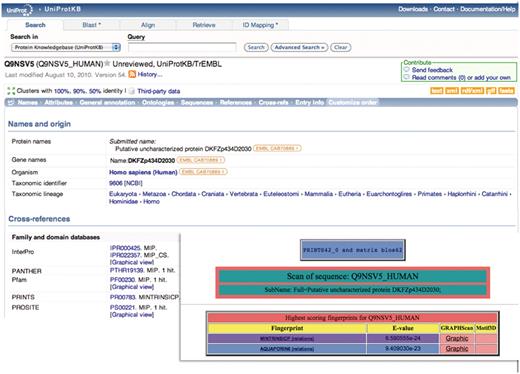
Illustration of a hierarchical PRINTS diagnosis. The UniProtKB/TrEMBL entry Q9NSV5_HUMAN was annotated as putative uncharacterized protein DKFZp434D2030; the family- and domain-database cross-references suggested membership of the major intrinsic protein (MIP) superfamily, but provided no specific family affiliation. The FingerPRINTScan result (inset) diagnoses the sequence both as a member of the MIP superfamily and as an aquaporin 6 subtype.
Annotation tools
As a significant portion of the work involved in creating PRINTS entails literature searching and the addition, by hand, of family-specific information to each new entry, in recent years we have developed a number of assistant tools to facilitate the annotation process. An early, simplistic approach, PRECIS (22), derived protein reports directly from UniProtKB/Swiss-Prot annotation, where possible detailing protein structure, function and disease associations, keywords, and database and literature cross-references. Later, we extended the method to exploit the scientific literature, using template- (23) and support vector machine (SVM)-driven (24) sentence-classification systems to extract pertinent sentences from PubMed abstracts.
Although useful for specific tasks, these tools were not designed to work together as a coherent package; their inputs and outputs were therefore different. For greater ease of use, we recently bundled together components of these systems, exploiting their most useful features to create an integrated Web-based annotator-assistant tool, termed MINOTAUR (25). To try to meet the needs of different users, the software offers a range of sequence- and text-based input options: (i) lists of UniProtKB/Swiss-Prot identifiers can be supplied in order to rapidly generate a basic PRECIS report; (ii) individual sequences may be input to a BLAST process, which runs behind the scenes and then automatically generates a PRECIS report from UniProtKB/Swiss-Prot sequences matching above the significance threshold; (iii) keyword queries may be used to cull relevant abstracts from PubMed in order to seed down-stream sentence extraction—suitable queries may also be suggested directly by the software in response to inputs (i) and (ii) above; alternatively, (iv) users may upload their own text corpus in PubMed XML format, again for downstream sentence processing. For options (iii) and (iv), the system gathers and processes the returned abstracts (or input text), allowing users to rank them according to relevance and/or to extract pertinent sentences using SVM- and rule-based sentence-classification systems (relating to structure, function, disease association, tissue specificity and subcellular localization). User-selected sentences may then be collated, with their relevant literature citations, into formatted paragraphs, which can be downloaded into text files to facilitate annotation tasks.
To illustrate how the MINOTAUR assistant tool can be used to derive functional insights for individual query sequences, Figures 2–4 show the results of searches using Q9C929_ARATH and Q30HW6_9CICH as queries. In the first example, UniProtKB describes Q9C929_ARATH as a putative G-protein-coupled receptor (GPCR); however, the entry contains cross-references to protein family and domain-based databases that suggest a relationship with the lanthionine synthetase component (LanC)-like proteins. If we use this sequence to seed PRECIS via the BLAST input option (Figure 2a), a report is rapidly created in which the cited literature (culled automatically from its six closest homologues, using default parameters) unambiguously points to a relationship with the LanC-like, rather than the GPCR, protein family; this is corroborated independently both by cross-references to family diagnoses from databases such as PRINTS and InterPro, and by references to the 3D structure determination (Figure 2b).
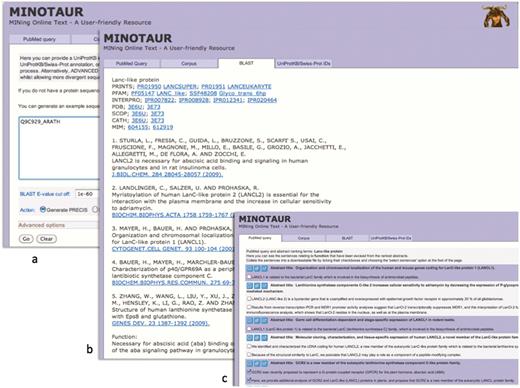
Using the MINOTAUR curator-assistant tool to generate a protein report and extract structure-related sentences from the literature: (a) shows the BLAST-PRECIS input option, with putative G-protein-coupled receptor, Q9C929_ARATH, as the query sequence; (b) shows the returned PRECIS report from the top 7 BLAST hits, which suggests the sequence really belongs to the LanC-like protein family; (c) selection of relevant sentences from the PubMed query results, confirming that the sequence is unlikely to be a GPCR.
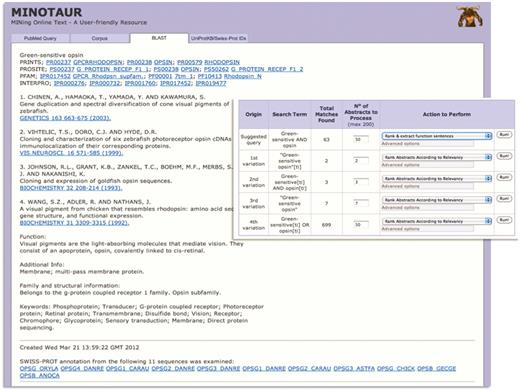
Using MINOTAUR to generate a PRECIS report for query sequence, Q30HW6_9CICH. The report, culled from the top 11 BLAST hits, suggests the sequence is a green-sensitive opsin—accordingly, the annotation extracted from UniProtKB/Swiss-Prot relates to the function of opsins. However, the hierarchical PRINTS diagnosis suggests that the sequence is a rhodopsin. To shed light on the discrepancy, the sequence can be used to generate possible PubMed queries—the inset shows that the recommended query is ‘Green-sensitive AND opsin’, and the SVM-based ‘rank and extract function sentences’ qualifier has been selected to extract sentences from the 63 retrieved abstracts.

Using MINOTAUR to select function-related sentences relevant to query sequence, Q30HW6_9CICH. The top sentences are shown, following use of the search options illustrated in Figure 3. The parent abstracts for each group of sentences may be quickly viewed by clicking on the appropriate icon (inset). In the examples highlighted, a set of green-sensitive opsins is noted to belong to a distinct ‘rhodopsin-like’ phylogenetic group, being more similar to rhodopsins than they are to other green pigments. This helps to resolve the apparent ambiguity in the PRINTS cross-reference to rhodopsins rather than to green-sensitive opsins: sequences in this group clearly have a rhodopsin-like sequence signature and not a ‘green’ one.
To shed light on this disparity by seeking further relevant literature, the same sequence can be used to generate possible PubMed queries. The query recommended by the software is ‘LanC-like AND protein’; running this with the SVM-based ‘rank and extract structure sentences’ qualifier generates a number of sentences from nine possible abstracts. As shown in the figure, sentences from the retrieved articles explain that the sequence is unlikely to be a bona fide GPCR because it does not contain the canonical seven transmembrane domains, and is more likely to be a LanC homologue (26)—for maximum flexibility, the software allows these supporting sentences to be selected (Figure 2c) and downloaded in order to augment the PRECIS report.
The second example is more subtle. UniProtKB describes Q30HW6_9CICH as a putative green-sensitive visual pigment; however, the entry contains cross-references to protein family and domain-based databases with conflicting annotation, suggesting that, rather, the sequence is a rhodopsin—interestingly, there is no cross-reference to the green-sensitive fingerprint in PRINTS. If we use the sequence to seed PRECIS via the BLAST input option (for convenience, using an e-value threshold of e−120 to reduce the number of processed sequences), the BLAST output reveals both green- and blue-sensitive opsins and rhodopsins among the top hits. The PRECIS report itself, while suggesting the protein is a green-sensitive opsin, also provides a cross-reference to the rhodopsin signature in PRINTS, suggesting again that the sequence is more similar to rhodopsins than it is to green-sensitive pigments—the fourth reference in the report [A visual pigment from chicken that resembles rhodopsin (27)] provides a further clue that perhaps there is something ‘odd’ about this sequence (Figure 3).
To glean further information from the literature, as before, the sequence can be used to generate possible PubMed queries (again, with the e-value threshold of e−120). The recommended query is ‘Green-sensitive AND opsin’; this can be run with the SVM-based ‘rank and extract function sentences’ qualifier (Figure 3, inset) to extract a range of sentences from the 63 possible abstracts (Figure 4). The retrieved sentences are highly relevant, and their parent abstracts may be quickly viewed by clicking on the appropriate icon (Figure 4, inset). In the examples highlighted, chicken, fish and lizard green-sensitive opsins are noted to belong to a distinct ‘rhodopsin-like’ phylogenetic group, being more similar to rhodopsins than they are to other green pigments, the functional and evolutionary consequences of which were investigated in detail by Kawamura and Yokoyama (28), and by Shichida et al. (29).
Analysis tools
The starting point for creating PRINTS entries involves creation and manual inspection of multiple sequence alignments. Conserved regions are selected by hand and used to assemble groups of motifs that uniquely differentiate closely related families from each other: the collection of motifs (the fingerprint) is scanned iteratively against a source sequence database (a UniProtKB/Swiss-Prot-TrEMBL composite) until no further new family members can be identified. At this point, the result is ready for manual annotation, prior to accession to PRINTS.
In an attempt to simplify this process, we have built many of these tasks into the core functionality of the evolving Utopia protein sequence analysis software suite (30, 31). Utopia permits both automatic and manual creation of alignments, and it allows motifs to be identified, selected, grouped as fingerprints and output for subsequent database searching. As Utopia has a modular architecture, it is readily customizable via Web-service ‘plugins’. We have therefore augmented the core functionality with a number of additional PRINTS-related tools. As illustrated in Figure 5, these include options to annotate groups of sequences within alignments (Figure 5a) via a PRECIS plugin (Figure 5b) and to search their constituent sequences directly against PRINTS via a FingerPRINTScan plugin (Figure 5c). For greater accessibility and to facilitate their use in other annotation projects, we added the WSDL files for these services to the EMBRACE Web-service registry (32, 33), whose core content now forms the backbone of the BioCatalogue (34).
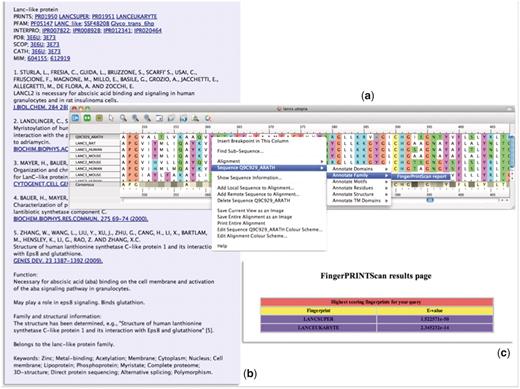
Illustration of the PRECIS and FingerPRINTScan Web-service plugins integrated within Utopia’s CINEMA alignment editor: (a) shows an alignment of sequence Q9C929_ARATH with LanC-like proteins, with the context-sensitive menu invoking a Web-service plugin; (b) shows the report generated for this group of sequences by the PRECIS plugin; and (c) shows the FingerPRINTScan-plugin result for a PRINTS search with Q9C929_ARATH, which diagnoses the sequence as a eurkaryotic LanC-like protein belonging to the LanC-like superfamily.
Database and software availability
For local installation, PRINTS flat-files and support files for its search tools are available from the anonymous-ftp server at Manchester (ftp.bioinf.man.ac.uk/pub/prints/)—the flat-files may also be retrieved from the EBI (ftp.ebi.ac.uk/pub/databases/prints) and NCBI (ncbi.nlm.nih.gov/repository/PRINTS). The database is accessible for online search and interrogation at www.manchester.ac.uk/dbbrowser/PRINTS/; MINOTAUR is accessible from www.bioinf.manchester.ac.uk/dbbrowser/minotaur/; the Utopia visualization and analysis tools may be downloaded for local installation from utopia.cs.man.ac.uk/utopia/; and the WSDL files are accessible from www.biocatalogue.org.
Discussion and conclusion
To date, 2156 fingerprints have been developed, manually annotated and deposited in PRINTS. Like PROSITE (release 20.79 of which documents 1632 entries) (35), the growth of the database remains slow, detailed annotation of entries being the rate-determining step; however, the extent of their manually crafted annotations sets these resources apart from those that exploit greater levels of automation, for which there is often limited or no biological documentation and limited validation, or in which family groupings may change between database releases.
While family- and domain-based databases that exploit significant amounts of automation in their production pipelines clearly benefit from more comprehensive coverage of sequence space, this can come with significant accuracy trade-offs. For example, Wong et al. (36) reported more than 1000 problems with Pfam domains: here, inclusion in the derived hidden Markov models of signal peptides and small numbers (usually 1–4) of transmembrane domains led to matches with proteins having nothing in common with the domain except for the occurrence of a hydrophobic region, presumably owing to selective pressures of the physical requirements of the membrane environment rather than homology. Schnoes et al. also reported a range of enzyme mis-annotations in public databases, where general, high-level annotation of promiscuous domains or broad superfamilies have been taken as proxies for the specific functions of matching sequences, failing to make the correct subfamily assignment and hence leading function annotation astray (37).
Naturally, the manually derived motif-based databases like PROSITE and PRINTS also contain errors. However, these are mitigated to some extent both by the greater selectivity of the methods they employ, and by the declaration within the databases of all known false-positive matches. In databases where this is not done, this may consequently give users the illusion that all of the gathered hits are ultimately correct (36). This has important down-stream consequences. These databases are commonly used in genome-/proteome-wide annotation projects—any errors they contain therefore tend to percolate (37, 38) to primary sequence archives like UniProtKB/TrEMBL and GenBank NR (39). The rising, largely unquantified error rates now recognized as infecting both protein sequence and protein family databases highlights an awkward tension between the necessity of deploying increasing levels of automation in functional annotation processes to cope with the inexorable growth of available sequence data, and the concomitant need for greater levels of manual scrutiny (37).
Mindful of this tension, we provide PRINTS not as an up-to-date match look-up table that tracks the current version of UniProt (this role is performed by InterPro) but, rather, as a manually fine-tuned diagnostic complement of the portfolio of protein family- and domain-based databases now available to sequence analysts. Keen to facilitate manual-annotation processes, we have developed decision-support rather than fully-automated curator-assistant software. The most recent, MINOTAUR, blends a range of existing tools and algorithms (BLAST, PRECIS, SVMs, etc.) with human interaction, bringing traditional sequence-analysis and literature-mining tools together within a single interface. Importantly, this allows rapid, automatic generation of protein reports from existing information in UniProtKB/Swiss-Prot and their manual enhancement with relevant sentences extracted directly from the biomedical literature.
In the first example presented above to illustrate the use of MINOTAUR, starting with a single sequence identifier with an erroneous UniProtKB description, it was possible swiftly and easily to garner sufficient information both from the literature and from a variety of protein-related databases to make the correct diagnosis. Of course, a straightforward BLAST search also quickly suggests the relationship of Q9C929_ARATH with the LanC-like proteins; the benefit of using MINOTAUR here is that it melds together in a brief report the UniProtKB/Swiss-Prot annotation from the top-matching BLAST sequences, and allows the user to exploit this both to challenge the UniProtKB/TrEMBL description and to explore relevant literature to find independent supporting articles with which to enhance the report.
In the second example, for a sequence with a ‘putative’ functional assignment and apparently conflicting protein family database cross-references, the PRECIS report, coupled with the literature automatically culled by the software, quickly offered supporting evidence for the UniProtKB functional assignment and resolved the ambiguity of what at first glance appeared to be an incorrect PRINTS cross-reference to rhodopsins rather than green pigments. For PRINTS and other database curators whose work is largely manual, and who cannot have expert knowledge of every protein family, tools of this type hence have the potential to help resolve ambiguities and correct errors, and ultimately to significantly ease their annotation burdens.
While increasing dependence on automation contributes to mounting error rates in our databases, manual efforts like PRINTS remain useful. As such, the database continues to play an important role, augmenting InterPro with deep family hierarchies and substantial annotation. The database and its related search, annotation and visualization tools are freely accessible both via the Web and as an integral part of the Utopia desktop application, thereby helping to improve its effectiveness as a fine-grained instrument for protein sequence analysis and genome-/proteome annotation.
Funding
European IMPACT project (contract number 213037 to A.M. and P.P.); the European FP6 EMBRACE project (contract number LHSG-CT-2004-512092 to A.C.); EuroKUP COST Action (BM0702 to A.T., I.P., A.T.).
Conflict of interest. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}