





Abstract
BioC is a new simple XML format for sharing biomedical text and annotations and libraries to read and write that format. This promotes the development of interoperable tools for natural language processing (NLP) of biomedical text. The interoperability track at the BioCreative IV workshop featured contributions using or highlighting the BioC format. These contributions included additional implementations of BioC, many new corpora in the format, biomedical NLP tools consuming and producing the format and online services using the format. The ease of use, broad support and rapidly growing number of tools demonstrate the need for and value of the BioC format.
Database URL:http://bioc.sourceforge.net/
Introduction
A vast amount of biomedical information is available as free text. But this information is available in a bewildering array of distinct formats. Adapting tools to each format is tedious and makes no direct contribution. BioC is a response to this situation ( 1 ).
BioC is a simple format for text, annotations on that text and relations between those annotations and other relations. It also includes libraries for reading data into and writing data out of native data structures in a number of common programming languages ( 2 ). These libraries allow tool developers to concentrate on the desired task and goal, largely ignoring the input or output format.
XML was chosen as the base format for BioC because it is well known and well documented. Standard XML tools can be used when appropriate and convenient. XML can handle the character sets and encodings in which biomedical text can be found. In addition to text passages, BioC uses standoff annotations to indicate particular portions of the text that are of interest. These annotations can be linguistic, such as parts of speech or syntactic structures, or they can be biological, such as disease or gene names. Standoff annotations are separate from the original text, leaving it unchanged, unlike in-line annotations. Standoff annotations can overlap or nest, however necessary, without conflict. Finally, many annotations are related to each other. Relation elements indicate which annotations are related and what role each particular annotation plays in the relation. Relations may be simple, such as indicating which abbreviation definition corresponds to a particular abbreviation, or they can be nested and complex such as protein–protein interaction events.
BioC implementations define native language data structures to hold the BioC information. Then developers can use native language data structures or objects they are comfortable with to access the BioC text, annotations and relations. Data are read from XML to the data classes and written from the data classes to XML using connector classes. These connector classes wrap standard XML parsers so they are robust and reliable. The developers can largely ignore the fact that their data reside in XML and concentrate on using the data in their native language data structures.
The BioC workflow is organized as described in Figure 1 . After the data are read into the BioC data classes, any needed processing can be performed. When that work is complete, the results are stored in BioC data classes and then written out in the BioC format. The separation between the BioC input/output code and the algorithms’ implementation is intentional. This structure makes it easier to adapt existing programs and leads to easier-to-modify programs.
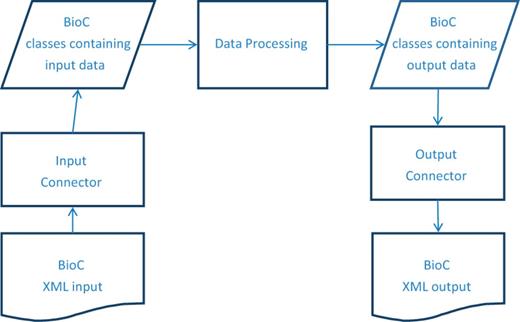
BioC process sequence. The BioC workflow allows data in the BioC format, from a file or any other stream, to be read into the BioC data classes via the Input Connector, or written into a new stream, via the Output Connector. The Data Processing module stands for any kind of NLP or text mining process that uses these data. Several processing modules may be chained together between input and output.
Communicating the precise information available in a file, and the tags or labels used to indicate this information, is an important part of data exchange. BioC uses key files to communicate this information. A key file is a plain text document composed by the author of the corpus explaining important organizing details and giving the meaning of tags or tag sets used in the data. A key file typically includes the character set and encoding, the entities annotated and, if the entities have been normalized, the ontology or controlled vocabulary used. A simple example of a BioC file appears in Figure 2 and the corresponding BioC key file is in Figure 3 . Initially, developers have a lot of flexibility setting up this information. As the community matures, consensus will lead to standards. Prior examples should be followed unless a new type of information is being shared.

Simple example of a BioC file.

An important benefit of a common format is tool interoperability. Many tools were originally developed, each with a particular format in mind. For tools using different formats to work together, much effort is required to modify one or both tools. Using a common format eliminates this barrier to integration. The interoperability track at BioCreative IV allowed developers to gain experience with BioC. It also led to the creation of a number of tools and corpora to encourage even broader use and reuse of BioC data and tools.
Contributors to the BioC interoperability track were asked to prepare a BioC module that could be seamlessly coupled with other BioC tools, and that performed an important natural language processing (NLP) or BioNLP task. Immediately following this introduction is a brief summary of these contributions. They are organized by type: BioC implementations, downloadable BioC tools, online BioC compatible services and available corpora in the BioC format. More details are then shared by the contributing groups. That is followed by some suggestions for using BioC in a shared task ( 3 , 4 ). This overview concludes with some thoughts on future directions.
Summary
Implementations
A BioC implementation consists of both computer language structures to hold the BioC data and modules to read and write the data from and to XML. Implementations of BioC in C++ and Java were available before the workshop ( 1 ). As part of the workshop, several additional implementations were developed ( 2 ). Two of these BioC implementations are for Perl and Python using SWIG to wrap the C++ implementation. This approach has the advantage that now BioC can be easily extended to other languages supported by SWIG. A native Python implementation was also created, which, of course, has the advantage of behaving exactly as expected by Python developers. There is also an implementation in Go, the intriguing new language from Google ( http://golang.org/ ).
Developing a new BioC implementation is fairly straightforward and has two major steps. First, data structures or objects need to be developed to hold the information. These should be as simple as possible and follow the languages’ expected conventions for holding data. Second, an XML parser needs to be chosen. A good parser allows both the simplicity of placing all the data in memory immediately and the efficiency of only reading the data as needed. With these steps complete, developers can easily access and store their data in the BioC structures, and the input/output (I/O) to and from XML files will be transparent.
Tools
A number of tools using BioC can be downloaded and applied to local data or combined with existing processes. NLP often begins with a linguistic preprocessing pipeline. Two commonly used tool sets, the MedPost ( 5 ) and Stanford ( 6 ) NLP tool sets, have been adapted to process text in the BioC format ( 7 ). These pipelines include tools such as sentence segmentation, tokenization, lemmatization, stemming, part-of-speech tagging and parsing. One advantage offered by the BioC format is that these tools can be mixed and matched, regardless of whether the researcher is working in C++, Java or another language with BioC support.
Abbreviation definition tools implementing three different algorithms are available in BioC ( 8 ). Schwartz and Hearst ( 9 ) is a well-known, simple and surprisingly effective algorithm. Ab3P uses a rule-based approach ( 10 ). These rules were developed using an approximate precision measure and are adapted to the length of the abbreviation. NatLAb was developed using machine learning on a naturally labeled training set using potential definitions and random analogs ( 11 ).
A number of named entity recognition (NER) tools are available in the BioC format ( 12 ). These include DNorm for disease names ( 13 , 14 ), tmVar for mutations ( 15 ), SR4GN for species ( 16 ), tmChem for chemicals ( 17 ) and GenNorm for gene normalization ( 18 ). The results of these tools can be used directly or as features for even further entity recognition or understanding tasks. In addition, PubTator, a web-based annotation tool, has also been adapted to BioC ( 19 ).
Another frequently used format in the biomedical community is brat rapid annotation tool (brat) ( http://brat.nlplaborg/standoff.html ) ( 20 ). The Brat2BioC tool allows two-way conversion between brat and BioC ( 21 ). This allows researchers to intermingle resources in either format.
Services
A number of online services have been made available. Argo ( 22 ) is a web-based text mining platform. Workflows on the platform can now use the BioC format ( 23 ). Example workflows include extraction of biomolecular events, identification of metabolic process concepts and recognition of Comparative Toxicogenomics Database (CTD) concepts.
Semantic role labeling (SRL) is an important task for recovering information about biological processes. BIOmedical SeMantIc roLe labEler (BIOSMILE) offers an online SRL service for files formatted in BioC ( 24 ).
One of the challenges of applying NLP tools to biomedical text is the complicated sentence structures typically used. Sentence simplification tools, such as iSimp, transform complicated sentences into sentences easier to comprehend and process. iSimp is available as a web service that processes BioC formatted text files ( 25 ).
Many teams contributed web-based NER tools using the BioC format for the CTD triage task. Using a common format was an important consideration for making the tools practical and useful to the CTD project ( 4 , 26 ).
Corpora
A format without data in that format is just an idea. There are now a number of corpora available in the BioC format. Some were explicitly prepared in the BioC format for this workshop. Others were used to train or develop the tools or services mentioned above. The rest demonstrate the results of an available tool or service. Most of these corpora use PubMed®, a collection of biomedical literature citations, or PubMed Central® (PMC) text, a free archive of full-text biomedical and life sciences journal literature. Any of these corpora can be used for the development and analysis of new biomedical NLP methods and techniques. A regularly updated list of BioC formatted corpora is maintained at the BioC Web site ( http://bioc.sourceforge.com ).
A significant contribution is the conversion of many corpora in the Wissensmanagement in der Bioinformatik (WBI) repository to the BioC format ( http://corpora.infor matik.hu-berlin.de/ ). Corpora in this repository include genes, mutations, chemicals, protein–protein interactions, disease–treatment relations and gene expression and phosphorylation events. Brat2BioC was also used to make the Human Variome ( 27 ) and CellFinder ( 28 ) corpora available in BioC.
The NCBI disease corpus of hand-annotated disease names is now available in the BioC format ( 29 ). In addition, it was processed by the C++ and Java pipelines, so it has available a number of linguistic annotations.
The Schwartz and Hearst ( 9 ) and Ab3P ( 10 ) abbreviation detection algorithms were accompanied by corpora developed to measure their performance. These corpora have been converted to BioC. Two additional abbreviation identification corpora, Medstract ( 30 ) and BIOADI ( 31 ), have also been converted to BioC ( 8 ).
The CTD developers have made available an annotated sample corpus for their workshop track ( 32 ). Likewise, the Gene Ontology (GO) task developers provide the BC4GO corpus, which has GO annotations and supporting sentences in the BioC format ( 33 ). Argo-related resources available in BioC include the Metabolites corpus ( 34 ). Finally, the iSimp corpus demonstrates examples of simplified sentences ( 35 ).
Contributions
This section provides an overview of the individual contributions to the BioC interoperability track.
Karin Verspoor and Antonio Jimeno Yepes
Translation of commonly used annotation formats into BioC allows reuse of existing annotated corpora with BioC solutions. The standoff brat format ( http://brat.nlplab.org/standoff.html ) is a commonly used format ( 20 ). For instance, it has been used in the BioNLP shared task series for annotated training data ( 36–38 ). Several recent biomedical corpora have been made available in the brat format, including the Human Variome Project corpus ( 27 ) and the CellFinder corpus ( 28 ). We have developed a software solution, named Brat2BioC ( 21 ), to translate annotations specified in brat format into BioC and vice versa. The Brat2BioC tool is available in Bitbucket at https://bit bucket.org/nicta_biomed/brat2bioc . Several differences exist between the BioC and brat formats. These include the physical division of data and annotations among various files, and the representational choices for entity and relation annotations. We have proposed and implemented resolutions for these differences to perform the mapping between the two formats.
This paragraph reports the detailed decisions made when converting between brat and BioC and may be of interest to only those with knowledge of both formats. The set of document files from the source brat corpus are converted to a single BioC file in our implementation. The identifier of each generated BioC document is the name of the source brat document file without the txt extension. We capture brat file extensions through an infon object that specifies the extension of the source file in which the annotation was found (a1, a2 or ann) instead of maintaining separate files for each source extension. We have modeled one brat document as one BioC passage, and no assumptions were made about the semantics of line breaks in the original brat file. With respect to annotation types, brat provides several annotation types that need to be mapped to the BioC annotation and relation entities. Specific infon tags for each brat annotation type have been used to cover this variety. Brat2BioC was used to convert a large set of corpora that are available for download and visualization from the WBI repository, as discussed below. With the application of Brat2BioC, the corpora in that repository are now available in both brat and BioC format.
Mariana Neves
Gold standard corpora are important resources for both the development and evaluation of new methods in the biomedical NLP domain. They provide means to train supervised learning systems and to carry out a fair comparison among different solutions under the same conditions. Hence, an important contribution to the BioC initiative, to help its adoption by this community, is the availability of existing corpora in this format. During participation in the BioC task in the BioCreative IV, most of the corpora that are available in the WBI repository ( http://corpora.infor matik.hu-berlin.de/ ) were converted to the BioC format. The repository currently contains >20 biomedical corpora whose annotations range from named-entities (e.g. genes/proteins, mutations, chemicals) and binary relationships (e.g. protein–protein interactions, disease–treatment relations) to biomedical events (e.g. gene expression, phosphorylation). Examples of corpora included in the repository are the following: AIMed, BioInfer, BioText, CellFinder, Drug-Drug Interaction Extraction 2011, Drug-Drug Interaction Extraction 2013, GeneReg, GENIA, GETM, GREC, HPDR50, IEPA, LLL, OSIRIS, SNP Corpus corpora and Variome. A complete list and a description of each corpus are provided on the web page. Originally, the repository was created to allow online visualization of biomedical corpora using the stav/brat annotation tool ( http://brat.nlplab.org/embed.html ). Since our participation in the BioC task, it also provides download functionality in the BioC format for the corpora whose license allows their redistribution. Conversion was carried out using the Brat2BioC conversion tool (cf. previous section), which allows conversion of corpora from the brat standoff format to the BioC format. An important next step regarding these corpora is a careful analysis and normalization of the entity and relationship types, as different corpora refer to the same concept using different names, e.g. ‘gene’, ‘protein’ and ‘GeneProtein’ for gene/proteins annotations.
Hernani Marques and Fabio Rinaldi
In BioCreative IV, Track-1 participants were asked to contribute to the BioC community in the area of interoperability. The Ontogene team based in Zurich noticed that no native BioC library for use with the Python programming language was available. We took this opportunity to create a Python implementation of the BioC library.
The PyBioC library recreates the functionality of the already available libraries in C++ or Java. However, we adhere to Python conventions where suitable, for example, refraining from implementing getter or setter methods for internal variables of the classes provided in PyBioC.
Basically the library consists of a set of classes representing the minimalistic data model proposed by the BioC community. Two specific classes (BioCReader and BioCWriter) are available to read in data provided in (valid) XML format and to write from PyBioC objects to valid BioC format. Validity is ensured by following the BioC DTD publicly available.
The library is being released with a BSD license and is available on a public github repository ( https://github.com/2mh/PyBioC ). The repository includes example programs. One sample program simply reads in and writes to BioC format. Another can tokenize and stem a BioC input file using the Natural Language Toolkit library ( http://nltk.org/ ). These examples are in the src directory of the distribution.
The OntoGene team has additionally used BioC as I/O formats for web services implemented within the context of their participation in the CTD task of BioCreative 2013. Currently, they are including PyBioC in their OntoGene pipeline with the aim of allowing remote access to its text mining capabilities.
PyBioC enables the biomedical text mining community to deal with BioC XML documents using a native implementation of the BioC library in the Python programming language. The authors welcome further contributions and additions to this work.
Hong-Jie Dai and Richard Tzong-Han Tsai
SRL is an important technique in NLP, especially for life scientists who are interested in uncovering information related to biological processes within literature. As a BioC contributor in the BioCreative IV interoperability track, we have developed a unique BioC module, which provides semantic analysis of biomedical abstracts to extract information related to location, manner, timing, condition and extent.
The BioC module BIOSMILE is an augmentation of our previous biomedical SRL system ( 39 ) developed under the BioProp standard and corpus ( 40 ). The BioC-BIOSMILE module can automatically label 30 predicates and 32 argument types. The predicates are collected from PubMed-indexed biomedical literatures and selected according to the frequency of usage. A total of 32 argument types are manually defined as location, manner, temporal, etc. Please refer to http://bws.iis.sinica.edu.tw/bioprop for more details.
BioC-BIOSMILE allows clients to submit one or more articles in the BioC format, and the server will return the SRL results in the BioC format. Tokenization and syntactic full parse tree structure information will be automatically generated by several NLP components, and the SRL results are returned accordingly. Further interpretation of the results is not necessary because the SRL annotations based on the parse tree are linked to phrases and tokens in the original sentences, which are returned to the client in the BioC format.
We believe that our module can support biomedical text mining researchers in developing or improving their systems. For example, in our previous work ( 41 ), we have integrated the SRL results in a PubMed-based online searching system. As for relation extraction tasks, such as protein–protein interaction or biomedical event extraction, the semantic role outputs of BioC-BIOSMILE can be encoded as features for machine learning models or in rules for pattern-based approaches.
BioC-BIOSMILE is available at http://bws.iis.sinica.edu.tw/BioC_BIOSMILE/BioC_Module.svc/SRL , and the demonstration Web site is http://bws.iis.sinica.edu.tw/bioc_biosmile .
Rafal Rak and Riza Theresa Batista-Navarro
The National Centre for Text Mining ( http://www.nactem.ac.uk ) at the University of Manchester prepared BioC-compliant tools related to three biomedical information extraction tasks: the extraction of biomolecular events, the identification of metabolic process concepts and the recognition of concepts in the CTD. The tools can be accessed as web services as well as directly in the web-based text mining platform Argo ( 22 ) ( http://argo.nactem.ac.uk ). Argo allows users to create custom workflows (pipelines) from the built-in library of elementary analytics that range from data serializers/deserializers to syntactic and semantic analytics to user-interactive components. Integration with third-party BioC-compliant modules is realized by the availability of BioC format reader and writer components, capable of deserializing and serializing BioC collections supplied as files (stored in users’ document spaces) or as web service end points. As a proof of concept and a tutorial for users, the authors created example workflows in Argo that perform the three aforementioned tasks. The workflows for the identification of metabolic process concepts and the recognition of concepts in CTD have been used in BioCreative IV’s Interactive Text Mining and CTD tracks (Rak et al ., in this special issue), respectively.
To complement the tools, the authors also transcribed several related resources, namely the Metabolites corpus ( 34 ) and a total of six biomolecular event corpora released for the BioNLP Shared Task 2011 ( https://sites.google.com/site/bionlpst/ ) and 2013 ( http://2013.bionlp-st.org ) series. These resources may be used in Argo to create comparative workflows, i.e. workflows that produce standard information retrieval performance metrics of a user-created workflow against one of the gold standard resources. Argo uses rich and well-defined annotation semantics facilitated by the adoption of the Unstructured Information Management Architecture ( 42 ) and, as such, complements the BioC format that defines only rudimentary semantics.
Yifan Peng and Cathy H. Wu
iSimp is a sentence simplification module designed to detect various types of simplification constructs and to produce one or more simple sentences from a given sentence by reducing its syntactic complexity ( 25 ). For example, from the complex sentence ‘Active Raf-2 phosphorylates and activates MEK1, which phosphorylates and activates the MAP kinases signal regulated kinases, ERK1 and ERK2 (PMID-8557975)’, iSimp produces multiple simple sentences, including ‘Active Raf-2 phosphorylates MEK1’, ‘MEK1 phosphorylates ERK1’, ‘MEK1 activates ERK1’ and so forth. We have demonstrated that this simplification can improve the performance of existing text mining applications ( 25 ).
iSimp adopts the BioC format( 35 ) to facilitate its integration into other text mining tools and workflows. The work contributes to (i) the development of a BioC tag set for annotating simplification constructs, (ii) a mechanism of using the BioC framework for denoting simplified sentences in a corpus file and (iii) the construction of several corpora in the BioC format for iSimp evaluation.
We define a BioC tag set for annotating and sharing the simplification results by using the annotation element to mark the simplification construct components and using the relation element to specify how they are related. In this way, we are able to assign roles for each component and skip over symbols like comma. Furthermore, we designed a unique schema for annotation of new simplified sentences. The BioC file thus generated contains both original and simplified sentences. While the offsets of the original sentences are the same as in the original text, those of the simplified sentences start with the next char after the last in the original document (offset of document + length of document). This new collection could then be treated as the input collection for further processing in an NLP pipeline. To evaluate the performance of iSimp, we constructed a BioC-annotated corpus, consisting of 130 Medline abstracts annotated with six types of simplification constructs. In addition, we converted the GENIA Event Extraction corpora of the BioNLP-ST 2011 ( 37 ) to BioC format to evaluate the impact of iSimp in relation extraction tasks. All these corpora have been made publicly available for evaluating and comparing various simplification systems ( http://research.dbi.udel.edu/isimp/corpus.html ).
The performance and usability evaluation results show that iSimp can be integrated into an existing relation extraction system seamlessly and easily via the BioC framework and can significantly improve system performance in terms of both precision and recall.
In the future, we aim for full adoption of BioC for broad dissemination of resources developed by the text mining group at the University of Delaware and the Protein Information Resource ( http://proteininformationresource.org/iprolink ), including curated literature corpora and text mining tools.
Thomas C. Wiegers and Carolyn J. Mattingly
The CTD ( http://ctdbase.org ) is a free publicly available resource that seeks to elucidate understanding of the mechanisms by which drugs and environmental chemicals influence the biological processes, which affect human health ( 26 ). CTD's PhD-level biocurators review the scientific literature and manually curate chemical–gene/protein interactions, chemical–disease relationships and gene– disease relationships, translating the information into a highly structured computable format ( 43 ). This manually curated information is then integrated with other external data sets to facilitate development of novel hypotheses about chemical–gene–disease networks ( 26 ). CTD typically selects curation topics by targeting specific chemicals. Depending on the chemical, there are often many more relevant articles than can be realistically curated. Consequently, we developed and implemented a highly effective fully functional text mining pipeline to ensure that biocurators review only those articles that are most likely to yield curatable information ( 44 ). At the heart of the pipeline is a ranking algorithm that scores each article in terms of its projected suitability for curation with a document relevancy score (DRS); integral to the algorithm are third party NER tools adapted for CTD use, and integrated directly into the pipeline.
Given its importance to the curation process, CTD is continuously researching ways to improve the effectiveness of the scoring algorithm. The ‘BioCreative Workshop 2012’ Track I/Triage task was organized by CTD and focused on document triaging and ranking ( 45 ). Participants developed tools that ranked articles in terms of their curatability and identified gene/protein, chemical/drug and disease actors, as well as action terms that describe chemical interactions in CTD. Although tools developed in conjunction with the track were effective, their impact was limited by a lack of interoperability. The tools were written using a wide variety of technologies and within technical infrastructures and architectures that would not necessarily easily integrate into CTD's existing pipeline. One alternative to potentially mitigate NER-related interoperability and general integration issues is the use of web services; rather than integrating NER tools directly into the CTD text mining pipeline, web services provide the capability to make simple calls from CTD's asynchronous batch-oriented text mining pipeline to remote NER web services. This approach tends to be inherently simpler than direct pipeline integration because the technical details of the tools themselves are completely abstracted by the web service.
To test this concept, CTD organized BioCreative IV, Track 3. Track 3 participants were instructed to provide Representational State Transfer (REST)-compliant web services-based NER tools that would enable CTD to send text passages to their remote sites to identify gene/protein, chemical/drug, disease and chemical/gene-specific action term mentions. The design of the track was predicated on one essential requirement: although internally the sites could be radically different from one another, externally all sites should behave identically from a communications perspective and be completely interchangeable. It was therefore critical that sites use one standard form of high-level interprocess communications. As Track 3 tasks were being analyzed and designed by CTD staff, NCBI-led collaborators were concurrently and coincidentally working on the development of BioC. The more CTD learned about and participated in development of BioC, the more it became clear that BioC's simple, lightweight, flexible design, along with its planned support across multiple programming languages and operating environments, made it an extremely attractive vehicle for Track 3 high-level interprocess communications. The timely emergence of BioC, coupled with REST's XML-centric nature and other attractive design features, made a REST/BioC-compliant architecture well positioned for use by Track 3.
Twelve research groups participated in Track 3, developing a total of 44 NER-based web services. Details of the NER results are summarized elsewhere in this Database BioCreative IV Virtual Issue. BioC proved to be an extremely robust effective tool in standardizing high-level interprocess communications. The framework provided all the functionality required for Track 3, and did so in an unobtrusive fashion: the vast majority of the participants required little, if any, help from the organizers with respect to BioC, and there were few errors associated with the BioC XML returned from the web services. Avoiding application-specific interprocess communication frameworks will ease future implementation within CTD. This led to the success of Track 3 and demonstrates a new approach for the text mining community in general. The participants developed 44 platform-independent web services, spanning four continents, encompassing four major NER categories, with varying levels of recall and precision, all using BioC as an interoperable communication framework. Many are expected to remain freely available.
Looking forward, CTD plans to collaborate with the top-performing teams in the individual NER categories, integrating their tools into the CTD text mining pipeline. Testing will then be conducted to determine whether the integration of these tools improve DRS scoring effectiveness. CTD's use of BioC will be expanded, requiring added sophistication beyond that used for Track 3, including text/CTD controlled vocabulary translation, and spatial orientation within the text passages. BioC is designed to easily accommodate this added sophistication. If testing is successful, we will incorporate these tools into the CTD curation pipeline, using BioC as the communications backbone.
In the end, the tools developed for Track 3 provided a level of interoperability that would not have otherwise existed in the absence of BioC. The results of Track 3 underscores the extraordinary ability of web services, coupled with BioC, to abstract the complexity of underlying computational systems and free users to focus on performance, rather than on the technical characteristics of the respective tool's underlying syntax and architecture.
Ritu Khare and Zhiyong Lu
We have recently developed several text mining tools for automatically recognizing key biomedical concepts such as chemicals, diseases, genes, mutations and species from the scientific literature ( 46 , 47 ). Each tool accepts a PubMed or PMC full-text article as an input and returns the biomedical entities at either mention-level or at both mention and concept levels. More specifically, our toolkit includes the following: (i) DNorm ( 13 , 48 ), an open-source software tool to identify and normalize disease names from biomedical texts, (ii) tmVar ( 15 ), a machine learning system for mutation recognition, (iii) SR4GN ( 16 ), a species recognition tool optimized for the gene normalization task ( 49 ), (iv) tmChem ( 50 ), a machine learning-based NER system for chemicals and (v) GenNorm ( 18 ), a rule-based tool for gene normalization ( 51 ). We applied at least four of these tools to the entire set of articles in PubMed and integrated their results in PubTator ( 19 , 52 , 53 ), a newly developed web-based tool for assisting manual corpus annotation and biocuration. More recently, we developed the BC4GO corpus ( 54 ), which consists of 200 full-text articles along with their GO annotations and supporting sentence information. BC4GO is the official data set for the BioCreative IV Track-4 GO Task ( 34 ), which tackles the challenge of automatic GO annotation through literature analysis.
When our tools were first developed, different input and output formats (e.g. free text, PMC XML format, PubTator format, GenNorm format, CHEMDNER format) were used. To improve the interoperability of our text mining toolkit, we produced an updated version of the toolkit, which we have named tmBioC. In particular, we modified each tool by adding the BioC format as a new input/output option. Because all our tools are focused on concept recognition, we used a single key file for interpreting the input full-text articles/abstracts and the output articles/abstracts with annotations. For the BC4GO Corpus, the 200 full-text articles were converted from the PMC XML data format to the BioC format. Separate key files were created to describe the full-text articles and the annotation files with GO annotations.
Our experience shows that only minimal changes were required to repackage our tools with BioC and produce tmBioC. Also, reading and writing to BioC format was fairly straightforward, as the functions and classes are already provided in the BioC library. For each tool, the primary developers modified their respective tools and confirmed the simplicity and learnability of the BioC format. The single key file, used by our five concept recognition tools and PubTator, could also evolve as a standard key file for concept recognition and annotation tasks as recommended in ( 1 ) and ( 55 ).
The tmBioC toolkit is freely available ( http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/tmTools ) and ready to be reused by a wider community of researchers in text mining, bioinformatics and biocuration. Our tools, although developed in different programming languages such as Java, Perl and C++, are now capable of sharing their inputs/outputs with each other, without any additional programming efforts. They can now also interact with other state-of-the-art tools to build more powerful applications. For example, a modular text mining pipeline of various BioC compatible tools for NER, normalization and relationship extraction could be developed to build sophisticated systems, e.g. an integrative disease-centered system connecting the biological and clinical aspects, providing information from causes (gene–mutation–disease relationship) to treatment (drug–disease relationships) of diseases by mining unstructured text (biomedical literature, clinical notes, etc.) and structured resources (data sets released by research organizations and groups). In the future, we anticipate much broader usage of BioC-compatible tools, as further efforts are invested in publicizing BioC.
Donald C. Comeau, Rezarta Islamaj Doğan and W. John Wilbur
Implementations
Implementations of BioC in C++ and Java were available before the workshop ( 1 ). This work contributed BioC implementations in three additional languages. Two implementations build directly on the C++ implementation. They use SWIG ( http://www.swig.org/ ) to wrap the C++ implementation and create Perl and Python implementations. This approach has several advantages. Once one language has been implemented, it is relatively easy to implement additional languages. It is guaranteed to be compatible with the C++ implementation because it is built on the C++ implementation. It performs at the speed of the C++ implementation, for the same reason. However, it is not a native implementation; that may lead to some surprises. We observed that SWIG had put more effort into implementing functions and wrappers for Python than for Perl. Thus, the Python version felt a bit more native than the Perl version.
Go is an intriguing new systems language from Google. It quickly compiles to machine language, offers the convenience of garbage collection and has convenient in-language concurrency. The long-term use of Go for bioNLP, or for general use, is unknown, but the growth curve is promising ( http://www.google.com/trends/explore?q= golang #q=golang&cmpt=q ).
Abbreviation definition recognition
Abbreviations, their definitions and their use are important for understanding and properly processing biomedical text documents. Three tools for abbreviation definition detection using the BioC format are available. The first is the well-known Schwartz and Hearst algorithm ( 9 ). Although simple, it produces good results that are difficult to improve on. Ab3P ( 10 ) is a rule-based algorithm that does give better precision and recall than Schwartz and Hearst. The developers created a precision approximation that allowed them to compare rules on millions of examples without human review. The third algorithm available in BioC—NatLAb—( 11 ) used machine learning to learn more flexible rules than Ab3P. It improves recall, with a modest loss of precision.
Several corpora were used to train and test these abbreviation detection programs and are also available in BioC. These are the Schwartz and Hearst ( 9 ), Medstract( 30 ), Ab3P( 10 ) and BIOADI ( 31 ) corpora. One important enhancement to these corpora encouraged by the BioC format was the specification of the exact location of each identified abbreviation definition. Earlier versions of the corpora simply stated the abbreviation definition. In addition, the annotations were reviewed for consistency and difficult cases were discussed by four human annotators. As a result, the quality of the annotations has been improved.
NLP pipeline
The first pass of NLP processing typically consists of a few common steps: sentence segmenting, tokenizing, part-of-speech identification, etc. NLP preprocessing pipelines were created in both C++ and Java. The C++ tools are based on the MedPost tools: sentence segmenting, tokenizing and part-of-speech tagging ( 5 ). In addition there is a wrapper for the C&C dependency parser ( 56 ) ( http://svn.ask.it.usyd.edu.au/trac/candc ). Most of the Java tools are based on the Stanford tools: sentence segmenting, tokenizing, part-of-speech tagging, dependency parsing and syntactic parsing ( 6 ). In addition, BioLemmatizer is available for lemmatization ( 57 ). An advantage of BioC is that the C++ tools and the Java tools can be mixed and matched to suit a project’s needs. Although possible in principle with earlier versions, this is now reasonable and practical.
As a practical demonstration of these pipelines, both pipelines were applied to the NCBI disease corpus ( 29 ). First the corpus of manually curated disease mentions was converted to the BioC format. Then it was processed by both the C++ and Java pipelines. Now the corpus is available in the BioC format containing both human annotations for disease and tool annotations for linguistic features.
BioC and running a shared task
One of the reasons BioC was created was to ease the challenge of shared tasks. Too often, participants in shared tasks and community challenges spend significant time understanding the data format and modifying their in-house programs to correctly input the data. That time could be better spent focusing on the challenge task. BioC addresses this issue. This section discusses how a shared task can benefit by using BioC for the corpus, annotations and evaluations.
Corpus
As covered earlier, a significant number of corpora annotated with biological information are already available in the BioC format. Even if the existing annotations are not directly useful for a task, the underlying text might be appropriate. There are projects underway to make PubMed references and the Open Access PMC articles available in BioC. To see what is currently available, check the web page http://bioc.sourceforge.com .
If no existing corpus will meet the needs of the task, adapting one to the BioC format is not an onerous effort. The organizers performing this task once, is much better than every participant needing to convert the corpus to match their particular needs. If a tool to read a format is available, creating a conversion tool from that format to the BioC format is simple and straightforward. This process requires copying the data from existing data structures to BioC data structures. Then the BioC implementation will write the data to the proper BioC format.
An important decision for a corpus is the character set and encoding. BioC can support either ASCII or Unicode. A related practical question is what unit should be used by annotations for offsets and lengths. For ASCII, it obviously should be bytes. For Unicode we recommend code points. This is the unit most likely to be convenient for programs processing the text. Using byte offsets is tempting because it allows using programs developed for ASCII. But it requires extra steps, including knowing the encoding used by the XML library on behalf of the BioC wrapper. As mentioned earlier, a key file is important for recording and sharing the choices made so that the corpus and annotations can be understood and processed properly.
BioC is not concise because XML is not concise. Compression solves this problem. The repeated element names are exactly the kind of data easily handled by compression algorithms.
Annotations
Many tasks will involve text annotation, both machine-generated and manually produced. Although a number of annotated corpora are now available in BioC format, they may not be ideal for a shared task. If they have been seen before, they may not be a true test of an algorithm’s ability, or the task may investigate issues not addressed by existing corpora. In either case, several manual annotation tools are available that work in the BioC format. Examples include PubTator ( 19 , 52 , 53 ) and BioQRator ( 58 ). Pre and post-processing with Brat2BioC, the brat ( http://brat.nlplab.org ) can also be used to create a BioC corpus.
Evaluation
A generic evaluation tool would be useful for BioC. An option is BRAT-Eval ( https://bitbucket.org/nicta_biomed/brateval/ ), again using Brat2BioC to incorporate it into a BioC pipeline. However, it is unlikely for any generic tool to be able to address all situations. For example, are results scored by document, individual annotation or by groups of related annotations? Must the scored annotations exactly match the gold standard, or is a reasonable overlap adequate? Most evaluations involving relations may need to be task-specific. For example, to evaluate abbreviation definition detection a task-specific evaluation tool was created. It scores appropriate pairs of annotations, indicated by a relation, not individual annotations. Fortunately, it is straightforward to prepare an appropriate evaluation tool because all the data are available in the native data structures of one’s development language.
With a common simple format, it is now easy to release the evaluation tool to challenge participants. Even though final testing will be performed on a test set held back by the organizers, releasing the evaluation tool can still help the participants in their development. This reduces surprises when the final test set is scored.
Conclusion
BioC is well positioned to fulfill its promise. A significant number of corpora and tools are currently available. Additional resources continue to be developed. New areas of applicability are being investigated. Yet, there is more work to be done. A common collection of key files, each describing BioC details and best practice suggestions for a number of typical bioNLP tasks, would help ensure interoperability. There is no need to invent new BioC conventions when previously created BioC files with the same type of annotations have led the way. Creativity should be reserved for new applications and new algorithms.
An important type of biomedical text, not yet publicly addressed by BioC corpora, is clinical text. Conversations with people familiar with clinical text, its needs and its properties greatly encourage us that BioC is well suited for clinical text. In fact, some initial private trials have been successful, but nothing has yet been released publicly.
Everything mentioned here is available directly, or indirectly, through bioc.sourceforge.com. We look forward to a time when using BioC will be considered routine.
Acknowledgements
M.N. would like to thank Prof. Ulf Leser for hosting the WBI repository.
Funding
National Science Foundation [DBI-0850319] to The BioCreative IV Workshop; Intramural Research Program of the National Institutes of Health, National Library of Medicine to D.C.C., R.I.D., R.K., Z.L. and W.J.W; NICTA, which is funded by the Australian Government through the Department of Communications, and the Australian Research Council through the ICT Centre of Excellence Program to A.J.Y. and K.V. Funding for open access charge: Intramural Research Program of the National Institutes of Health, National Library of Medicine.
Conflict of interest . None declared.
References
Author notes
Citation details: Comeau,D.C., Batista-Navarro,R.T., Dai,H-.J., et al . BioC interoperability track overview. Database (2014) Vol. 2014: article ID bau053; doi:10.1093/database/bau053

{kind=link}
{kind=link}
{kind=link}