





Abstract
Gene Ontology (GO) annotation is a common task among model organism databases (MODs) for capturing gene function data from journal articles. It is a time-consuming and labor-intensive task, and is thus often considered as one of the bottlenecks in literature curation. There is a growing need for semiautomated or fully automated GO curation techniques that will help database curators to rapidly and accurately identify gene function information in full-length articles. Despite multiple attempts in the past, few studies have proven to be useful with regard to assisting real-world GO curation. The shortage of sentence-level training data and opportunities for interaction between text-mining developers and GO curators has limited the advances in algorithm development and corresponding use in practical circumstances. To this end, we organized a text-mining challenge task for literature-based GO annotation in BioCreative IV. More specifically, we developed two subtasks: (i) to automatically locate text passages that contain GO-relevant information (a text retrieval task) and (ii) to automatically identify relevant GO terms for the genes in a given article (a concept-recognition task). With the support from five MODs, we provided teams with >4000 unique text passages that served as the basis for each GO annotation in our task data. Such evidence text information has long been recognized as critical for text-mining algorithm development but was never made available because of the high cost of curation. In total, seven teams participated in the challenge task. From the team results, we conclude that the state of the art in automatically mining GO terms from literature has improved over the past decade while much progress is still needed for computer-assisted GO curation. Future work should focus on addressing remaining technical challenges for improved performance of automatic GO concept recognition and incorporating practical benefits of text-mining tools into real-world GO annotation.
Database URL:http://www.biocreative.org/tasks/biocreative-iv/track-4-GO/ .
Introduction
Manual Gene Ontology (GO) annotation is the task of human curators assigning gene function information using GO terms through reading the biomedical literature, the results of which play important roles in different areas of biological research ( 1–4 ). Currently, GO (data-version: 9 September 2013 used in the study) contains >40 000 concept terms (e.g. cell growth) under three distinct branches (molecular function, cellular component and biological process). Furthermore, GO terms are organized and related in a hierarchical manner (e.g. cell growth is a child concept of growth), where terms can have single or multiple parentage ( 5 ). Manual GO annotation is a common task among model organism databases (MODs) ( 6 ) and can be time-consuming and labor-intensive. Thus, manual GO annotation is often considered one of the bottlenecks in literature-based biocuration ( 7 ). As a result, many MODs can only afford to curate a fraction of relevant articles. For instance, the curation team of The Arabidopsis Information Resource (TAIR) has been able to curate <30% of newly published articles that contain information about Arabidopsis genes ( 8 ).
Recently, there is a growing interest for building automatic text-mining tools to assist manual biological data curation (eCuration) ( 9–20 ), including systems that aim to help database curators to rapidly and accurately identify gene function information in full-length articles ( 21 , 22 ). Although automatically mining GO terms from full-text articles is not a new problem in Biomedical Natural Language Processing (BioNLP), few studies have proven to be useful with regard to assisting real-world GO curation. The lack of access to evidence text associated with GO annotations and limited opportunities for interaction with actual GO curators have been recognized as the major difficulties in algorithm development and corresponding application in practical circumstances ( 22 , 23 ). As such, in BioCreative IV, not only did we provide teams with article-level gold-standard GO annotations for each full-text article as has been done in the past, but we also provided evidence text for each GO annotation with help from expert GO curators. That is, to best help text-mining tool advancement, evidence text passages that support each GO annotation were provided in addition to the usual GO annotations, which typically include three distinct elements: gene or gene product, GO term and GO evidence code.
Also, as we know from past BioCreative tasks, recognizing gene names and experimental codes from full text are difficult tasks on their own ( 24–27 ). Hence, to encourage teams to focus on GO term extraction, we proposed, for this task, to separate gene recognition from GO term and evidence code selection by including both the gene names and associated NCBI Gene identifiers in the task data sets.
Specifically, we proposed two challenge tasks, aimed toward automated GO concept recognition from full-length articles:
Task A: retrieving GO evidence text for relevant genes
GO evidence text is critical for human curators to make associated GO annotations. For a given GO annotation, multiple evidence passages may appear in the paper, some being more specific with experimental information while others may be more succinct about the gene function. For this subtask, participants were given as input full-text articles together with relevant gene information. For system output, teams were asked to submit a list of GO evidence sentences for each of the input genes in the paper. Manually curated GO evidence passages were used as the gold standard for evaluating team submissions. Each team was allowed to submit three runs.
Task B: predicting GO terms for relevant genes
This subtask is a step toward the ultimate goal of using computers for assisting human GO curation. As in Task A, participants were given as input full-text articles with relevant gene information. For system output, teams were asked to return a list of relevant GO terms for each of the input genes in a paper. Manually curated GO annotations were used as the gold standard for evaluating team predictions. As in Task A, each team was allowed to submit three runs.
Generally speaking, the first subtask is a text retrieval task while the second can be seen as a multi-class text classification problem where each GO term represents a distinct class label. In the BioNLP research domain, the first subtask is in particular akin to the BioCreative II Interaction Sentence subtask ( 24 ), which also served as an immediate step for the ultimate goal of detecting protein–protein interactions. Task A is also similar to the BioCreative I GO subtask 2.1 ( 22 ) and automatic GeneRIF identification ( 18 , 28–31 ). The second subtask is similar to the BioCreative I GO subtask 2.2 ( 22 ) and is also closely related to the problem of semantic indexing of biomedical literature, such as automatic indexing of biomedical publications with MeSH terms ( 32–35 ).
Methods
Corpus annotation
A total of eight professional GO curators from five different MODs—FlyBase ( http://flybase.org/ ); Maize Genetics and Genomics Database ( http://www.maizegdb.org/ ); Rat Genome Database ( http://rgd.mcw.edu/ ); TAIR ( http://www.arabidopsis.org/ ); WormBase ( http://www.wormbase.org/ )—contributed to the development of the task data. To create the annotated corpus, each curator was asked, in addition to their routine annotation of gene-related GO information, to mark up the associated evidence text in each paper that supports those annotations using a Web-based annotation tool. To provide complete data for text-mining system development (i.e. both positive and negative training data), curators were asked to select evidence text exhaustively throughout the paper ( 36 ).
For obtaining high-quality and consistent annotations across curators, detailed annotation guidelines were developed and provided to the curators. In addition, each curator was asked to practice on a test document following the guidelines before they began curating task documents. Because of the significant workload and limited number of curators per group, each paper was only annotated by a single curator.
Evaluation measures
Results
The BC4GO corpus
The task participants were provided with three data sets comprising 200 full-text articles in the BioC XML format ( 39 ). Our evaluation for the two subtasks was to assess teams’ ability to return relevant sentences and GO terms for each given gene in the 50 test articles. Hence, we show in Table 1 the overall statistics of the BC4GO corpus including the numbers of genes, gene-associated GO terms and evidence text passages. For instance, in the 50 test articles, 194 genes were associated with 644 GO terms and 1681 evidence text passages, respectively. We refer interested readers to ( 36 ) for a detailed description of the BC4GO corpus.
Overall statistics of the BC4GO corpus
| Curated data | Training set | Development set | Test set |
|---|---|---|---|
| Full-text articles | 100 | 50 | 50 |
| Genes in those articles | 300 | 171 | 194 |
| Gene-associated passages in those articles | 2234 | 1247 | 1681 |
| Unique gene-associated GO terms in those articles | 954 | 575 | 644 |
| Curated data | Training set | Development set | Test set |
|---|---|---|---|
| Full-text articles | 100 | 50 | 50 |
| Genes in those articles | 300 | 171 | 194 |
| Gene-associated passages in those articles | 2234 | 1247 | 1681 |
| Unique gene-associated GO terms in those articles | 954 | 575 | 644 |
Overall statistics of the BC4GO corpus
| Curated data | Training set | Development set | Test set |
|---|---|---|---|
| Full-text articles | 100 | 50 | 50 |
| Genes in those articles | 300 | 171 | 194 |
| Gene-associated passages in those articles | 2234 | 1247 | 1681 |
| Unique gene-associated GO terms in those articles | 954 | 575 | 644 |
| Curated data | Training set | Development set | Test set |
|---|---|---|---|
| Full-text articles | 100 | 50 | 50 |
| Genes in those articles | 300 | 171 | 194 |
| Gene-associated passages in those articles | 2234 | 1247 | 1681 |
| Unique gene-associated GO terms in those articles | 954 | 575 | 644 |
Team participation results
Overall, seven teams (three from America, three from Asia and one from Europe) participated in the GO task. In total, they submitted 32 runs: 15 runs from five different teams for Task A, and 17 runs from six teams for Task B.
Team results of Task A
Table 2 shows the results of 15 runs submitted by the five participating teams in Task A. Run 3 from Team 238 achieved the highest F 1 score in both exact match (0.270) and overlap (0.387) calculations. Team 238 is also the only team that submitted results for all 194 genes from the input of the test set. The highest recall is 0.424 in exact match and 0.716 in overlap calculations by the same run (Team 264, run 1), respectively. The highest precision is 0.220 in exact match by Team 238 Run 2 and 0.354 in overlap by Team 183 Run 2. Also when evaluating team submissions using the relaxed measure (i.e. allowing overlaps), on average, the overlap between the text-mined and human-curated sentences was found to be >50% (56.5%).
Team results for Task A using traditional Precision (P), Recall (R) and F-measure (F1)
| Team | Run | Genes | Passages | Exact match | Overlap | ||||
|---|---|---|---|---|---|---|---|---|---|
| P | R | F 1 | P | R | F 1 | ||||
| 183 | 1 | 173 | 1042 | 0.206 | 0.128 | 0.158 | 0.344 | 0.213 | 0.263 |
| 183 | 2 | 173 | 1042 | 0.217 | 0.134 | 0.166 | 0.354 | 0.220 | 0.271 |
| 183 | 3 | 173 | 1042 | 0.107 | 0.066 | 0.082 | 0.204 | 0.127 | 0.156 |
| 237 | 1 | 23 | 54 | 0.185 | 0.006 | 0.012 | 0.333 | 0.011 | 0.021 |
| 237 | 2 | 96 | 2755 | 0.103 | 0.171 | 0.129 | 0.214 | 0.351 | 0.266 |
| 237 | 3 | 171 | 3717 | 0.138 | 0.305 | 0.190 | 0.213 | 0.471 | 0.293 |
| 238 | 1 | 194 | 2698 | 0.219 | 0.352 | 0.270 | 0.313 | 0.503 | 0.386 |
| 238 | 2 | 194 | 2362 | 0.220 | 0.310 | 0.257 | 0.314 | 0.442 | 0.367 |
| 238 | 3 | 194 | 2866 | 0.214 | 0.366 | 0.270 | 0.307 | 0.524 | 0.387 |
| 250 | 1 | 161 | 3297 | 0.146 | 0.286 | 0.193 | 0.239 | 0.469 | 0.317 |
| 250 | 2 | 140 | 2848 | 0.153 | 0.259 | 0.193 | 0.258 | 0.437 | 0.325 |
| 250 | 3 | 161 | 3733 | 0.140 | 0.311 | 0.193 | 0.226 | 0.503 | 0.312 |
| 264 | 1 | 167 | 13 533 | 0.052 | 0.424 | 0.093 | 0.088 | 0.716 | 0.157 |
| 264 | 2 | 111 | 2243 | 0.037 | 0.049 | 0.042 | 0.077 | 0.103 | 0.088 |
| 264 | 3 | 111 | 2241 | 0.037 | 0.049 | 0.042 | 0.077 | 0.103 | 0.088 |
| Team | Run | Genes | Passages | Exact match | Overlap | ||||
|---|---|---|---|---|---|---|---|---|---|
| P | R | F 1 | P | R | F 1 | ||||
| 183 | 1 | 173 | 1042 | 0.206 | 0.128 | 0.158 | 0.344 | 0.213 | 0.263 |
| 183 | 2 | 173 | 1042 | 0.217 | 0.134 | 0.166 | 0.354 | 0.220 | 0.271 |
| 183 | 3 | 173 | 1042 | 0.107 | 0.066 | 0.082 | 0.204 | 0.127 | 0.156 |
| 237 | 1 | 23 | 54 | 0.185 | 0.006 | 0.012 | 0.333 | 0.011 | 0.021 |
| 237 | 2 | 96 | 2755 | 0.103 | 0.171 | 0.129 | 0.214 | 0.351 | 0.266 |
| 237 | 3 | 171 | 3717 | 0.138 | 0.305 | 0.190 | 0.213 | 0.471 | 0.293 |
| 238 | 1 | 194 | 2698 | 0.219 | 0.352 | 0.270 | 0.313 | 0.503 | 0.386 |
| 238 | 2 | 194 | 2362 | 0.220 | 0.310 | 0.257 | 0.314 | 0.442 | 0.367 |
| 238 | 3 | 194 | 2866 | 0.214 | 0.366 | 0.270 | 0.307 | 0.524 | 0.387 |
| 250 | 1 | 161 | 3297 | 0.146 | 0.286 | 0.193 | 0.239 | 0.469 | 0.317 |
| 250 | 2 | 140 | 2848 | 0.153 | 0.259 | 0.193 | 0.258 | 0.437 | 0.325 |
| 250 | 3 | 161 | 3733 | 0.140 | 0.311 | 0.193 | 0.226 | 0.503 | 0.312 |
| 264 | 1 | 167 | 13 533 | 0.052 | 0.424 | 0.093 | 0.088 | 0.716 | 0.157 |
| 264 | 2 | 111 | 2243 | 0.037 | 0.049 | 0.042 | 0.077 | 0.103 | 0.088 |
| 264 | 3 | 111 | 2241 | 0.037 | 0.049 | 0.042 | 0.077 | 0.103 | 0.088 |
Both strict exact match and relaxed overlap measure are considered.
Team results for Task A using traditional Precision (P), Recall (R) and F-measure (F1)
| Team | Run | Genes | Passages | Exact match | Overlap | ||||
|---|---|---|---|---|---|---|---|---|---|
| P | R | F 1 | P | R | F 1 | ||||
| 183 | 1 | 173 | 1042 | 0.206 | 0.128 | 0.158 | 0.344 | 0.213 | 0.263 |
| 183 | 2 | 173 | 1042 | 0.217 | 0.134 | 0.166 | 0.354 | 0.220 | 0.271 |
| 183 | 3 | 173 | 1042 | 0.107 | 0.066 | 0.082 | 0.204 | 0.127 | 0.156 |
| 237 | 1 | 23 | 54 | 0.185 | 0.006 | 0.012 | 0.333 | 0.011 | 0.021 |
| 237 | 2 | 96 | 2755 | 0.103 | 0.171 | 0.129 | 0.214 | 0.351 | 0.266 |
| 237 | 3 | 171 | 3717 | 0.138 | 0.305 | 0.190 | 0.213 | 0.471 | 0.293 |
| 238 | 1 | 194 | 2698 | 0.219 | 0.352 | 0.270 | 0.313 | 0.503 | 0.386 |
| 238 | 2 | 194 | 2362 | 0.220 | 0.310 | 0.257 | 0.314 | 0.442 | 0.367 |
| 238 | 3 | 194 | 2866 | 0.214 | 0.366 | 0.270 | 0.307 | 0.524 | 0.387 |
| 250 | 1 | 161 | 3297 | 0.146 | 0.286 | 0.193 | 0.239 | 0.469 | 0.317 |
| 250 | 2 | 140 | 2848 | 0.153 | 0.259 | 0.193 | 0.258 | 0.437 | 0.325 |
| 250 | 3 | 161 | 3733 | 0.140 | 0.311 | 0.193 | 0.226 | 0.503 | 0.312 |
| 264 | 1 | 167 | 13 533 | 0.052 | 0.424 | 0.093 | 0.088 | 0.716 | 0.157 |
| 264 | 2 | 111 | 2243 | 0.037 | 0.049 | 0.042 | 0.077 | 0.103 | 0.088 |
| 264 | 3 | 111 | 2241 | 0.037 | 0.049 | 0.042 | 0.077 | 0.103 | 0.088 |
| Team | Run | Genes | Passages | Exact match | Overlap | ||||
|---|---|---|---|---|---|---|---|---|---|
| P | R | F 1 | P | R | F 1 | ||||
| 183 | 1 | 173 | 1042 | 0.206 | 0.128 | 0.158 | 0.344 | 0.213 | 0.263 |
| 183 | 2 | 173 | 1042 | 0.217 | 0.134 | 0.166 | 0.354 | 0.220 | 0.271 |
| 183 | 3 | 173 | 1042 | 0.107 | 0.066 | 0.082 | 0.204 | 0.127 | 0.156 |
| 237 | 1 | 23 | 54 | 0.185 | 0.006 | 0.012 | 0.333 | 0.011 | 0.021 |
| 237 | 2 | 96 | 2755 | 0.103 | 0.171 | 0.129 | 0.214 | 0.351 | 0.266 |
| 237 | 3 | 171 | 3717 | 0.138 | 0.305 | 0.190 | 0.213 | 0.471 | 0.293 |
| 238 | 1 | 194 | 2698 | 0.219 | 0.352 | 0.270 | 0.313 | 0.503 | 0.386 |
| 238 | 2 | 194 | 2362 | 0.220 | 0.310 | 0.257 | 0.314 | 0.442 | 0.367 |
| 238 | 3 | 194 | 2866 | 0.214 | 0.366 | 0.270 | 0.307 | 0.524 | 0.387 |
| 250 | 1 | 161 | 3297 | 0.146 | 0.286 | 0.193 | 0.239 | 0.469 | 0.317 |
| 250 | 2 | 140 | 2848 | 0.153 | 0.259 | 0.193 | 0.258 | 0.437 | 0.325 |
| 250 | 3 | 161 | 3733 | 0.140 | 0.311 | 0.193 | 0.226 | 0.503 | 0.312 |
| 264 | 1 | 167 | 13 533 | 0.052 | 0.424 | 0.093 | 0.088 | 0.716 | 0.157 |
| 264 | 2 | 111 | 2243 | 0.037 | 0.049 | 0.042 | 0.077 | 0.103 | 0.088 |
| 264 | 3 | 111 | 2241 | 0.037 | 0.049 | 0.042 | 0.077 | 0.103 | 0.088 |
Both strict exact match and relaxed overlap measure are considered.
Team results of Task B
Table 3 shows the results of 17 runs submitted by the six participating teams in Task B. Run 1 from Team 183 achieved the highest F 1 score in traditional (0.134) and hierarchical measures (0.338). The same run also obtained the highest precision of 0.117 in exact match while the highest precision in hierarchical match is 0.415 obtained by Run 1 of Team 237. However, this run only returned 37 GO terms for 23 genes. The highest recall is 0.306 and 0.647 in the two measures by Run 3 of Team 183.
Team results for the Task B using traditional Precision (P), Recall (R) and F1-measure (F1) and hierarchical precision (hP), recall (hR) and F1-measure (hF1)
| Team | Run | Genes | GO terms | Exact match | Hierarchical match | ||||
|---|---|---|---|---|---|---|---|---|---|
| P | R | F 1 | hP | hR | hF 1 | ||||
| 183 | 1 | 172 | 860 | 0.117 | 0.157 | 0.134 | 0.322 | 0.356 | 0.338 |
| 183 | 2 | 172 | 1720 | 0.092 | 0.245 | 0.134 | 0.247 | 0.513 | 0.334 |
| 183 | 3 | 172 | 3440 | 0.057 | 0.306 | 0.096 | 0.178 | 0.647 | 0.280 |
| 220 | 1 | 50 | 2639 | 0.018 | 0.075 | 0.029 | 0.064 | 0.190 | 0.096 |
| 220 | 2 | 46 | 1747 | 0.024 | 0.065 | 0.035 | 0.087 | 0.158 | 0.112 |
| 237 | 1 | 23 | 37 | 0.108 | 0.006 | 0.012 | 0.415 | 0.020 | 0.039 |
| 237 | 2 | 96 | 2424 | 0.108 | 0.068 | 0.029 | 0.084 | 0.336 | 0.135 |
| 237 | 3 | 171 | 4631 | 0.037 | 0.264 | 0.064 | 0.150 | 0.588 | 0.240 |
| 238 | 1 | 194 | 1792 | 0.054 | 0.149 | 0.079 | 0.243 | 0.459 | 0.318 |
| 238 | 2 | 194 | 555 | 0.088 | 0.076 | 0.082 | 0.250 | 0.263 | 0.256 |
| 238 | 3 | 194 | 850 | 0.029 | 0.039 | 0.033 | 0.196 | 0.310 | 0.240 |
| 243 | 1 | 109 | 510 | 0.073 | 0.057 | 0.064 | 0.249 | 0.269 | 0.259 |
| 243 | 2 | 104 | 393 | 0.084 | 0.051 | 0.064 | 0.280 | 0.248 | 0.263 |
| 243 | 3 | 144 | 2538 | 0.030 | 0.116 | 0.047 | 0.130 | 0.477 | 0.204 |
| 250 | 1 | 171 | 1389 | 0.052 | 0.112 | 0.071 | 0.174 | 0.328 | 0.227 |
| 250 | 2 | 166 | 1893 | 0.049 | 0.143 | 0.073 | 0.128 | 0.374 | 0.191 |
| 250 | 3 | 132 | 453 | 0.095 | 0.067 | 0.078 | 0.284 | 0.161 | 0.206 |
| Team | Run | Genes | GO terms | Exact match | Hierarchical match | ||||
|---|---|---|---|---|---|---|---|---|---|
| P | R | F 1 | hP | hR | hF 1 | ||||
| 183 | 1 | 172 | 860 | 0.117 | 0.157 | 0.134 | 0.322 | 0.356 | 0.338 |
| 183 | 2 | 172 | 1720 | 0.092 | 0.245 | 0.134 | 0.247 | 0.513 | 0.334 |
| 183 | 3 | 172 | 3440 | 0.057 | 0.306 | 0.096 | 0.178 | 0.647 | 0.280 |
| 220 | 1 | 50 | 2639 | 0.018 | 0.075 | 0.029 | 0.064 | 0.190 | 0.096 |
| 220 | 2 | 46 | 1747 | 0.024 | 0.065 | 0.035 | 0.087 | 0.158 | 0.112 |
| 237 | 1 | 23 | 37 | 0.108 | 0.006 | 0.012 | 0.415 | 0.020 | 0.039 |
| 237 | 2 | 96 | 2424 | 0.108 | 0.068 | 0.029 | 0.084 | 0.336 | 0.135 |
| 237 | 3 | 171 | 4631 | 0.037 | 0.264 | 0.064 | 0.150 | 0.588 | 0.240 |
| 238 | 1 | 194 | 1792 | 0.054 | 0.149 | 0.079 | 0.243 | 0.459 | 0.318 |
| 238 | 2 | 194 | 555 | 0.088 | 0.076 | 0.082 | 0.250 | 0.263 | 0.256 |
| 238 | 3 | 194 | 850 | 0.029 | 0.039 | 0.033 | 0.196 | 0.310 | 0.240 |
| 243 | 1 | 109 | 510 | 0.073 | 0.057 | 0.064 | 0.249 | 0.269 | 0.259 |
| 243 | 2 | 104 | 393 | 0.084 | 0.051 | 0.064 | 0.280 | 0.248 | 0.263 |
| 243 | 3 | 144 | 2538 | 0.030 | 0.116 | 0.047 | 0.130 | 0.477 | 0.204 |
| 250 | 1 | 171 | 1389 | 0.052 | 0.112 | 0.071 | 0.174 | 0.328 | 0.227 |
| 250 | 2 | 166 | 1893 | 0.049 | 0.143 | 0.073 | 0.128 | 0.374 | 0.191 |
| 250 | 3 | 132 | 453 | 0.095 | 0.067 | 0.078 | 0.284 | 0.161 | 0.206 |
Team results for the Task B using traditional Precision (P), Recall (R) and F1-measure (F1) and hierarchical precision (hP), recall (hR) and F1-measure (hF1)
| Team | Run | Genes | GO terms | Exact match | Hierarchical match | ||||
|---|---|---|---|---|---|---|---|---|---|
| P | R | F 1 | hP | hR | hF 1 | ||||
| 183 | 1 | 172 | 860 | 0.117 | 0.157 | 0.134 | 0.322 | 0.356 | 0.338 |
| 183 | 2 | 172 | 1720 | 0.092 | 0.245 | 0.134 | 0.247 | 0.513 | 0.334 |
| 183 | 3 | 172 | 3440 | 0.057 | 0.306 | 0.096 | 0.178 | 0.647 | 0.280 |
| 220 | 1 | 50 | 2639 | 0.018 | 0.075 | 0.029 | 0.064 | 0.190 | 0.096 |
| 220 | 2 | 46 | 1747 | 0.024 | 0.065 | 0.035 | 0.087 | 0.158 | 0.112 |
| 237 | 1 | 23 | 37 | 0.108 | 0.006 | 0.012 | 0.415 | 0.020 | 0.039 |
| 237 | 2 | 96 | 2424 | 0.108 | 0.068 | 0.029 | 0.084 | 0.336 | 0.135 |
| 237 | 3 | 171 | 4631 | 0.037 | 0.264 | 0.064 | 0.150 | 0.588 | 0.240 |
| 238 | 1 | 194 | 1792 | 0.054 | 0.149 | 0.079 | 0.243 | 0.459 | 0.318 |
| 238 | 2 | 194 | 555 | 0.088 | 0.076 | 0.082 | 0.250 | 0.263 | 0.256 |
| 238 | 3 | 194 | 850 | 0.029 | 0.039 | 0.033 | 0.196 | 0.310 | 0.240 |
| 243 | 1 | 109 | 510 | 0.073 | 0.057 | 0.064 | 0.249 | 0.269 | 0.259 |
| 243 | 2 | 104 | 393 | 0.084 | 0.051 | 0.064 | 0.280 | 0.248 | 0.263 |
| 243 | 3 | 144 | 2538 | 0.030 | 0.116 | 0.047 | 0.130 | 0.477 | 0.204 |
| 250 | 1 | 171 | 1389 | 0.052 | 0.112 | 0.071 | 0.174 | 0.328 | 0.227 |
| 250 | 2 | 166 | 1893 | 0.049 | 0.143 | 0.073 | 0.128 | 0.374 | 0.191 |
| 250 | 3 | 132 | 453 | 0.095 | 0.067 | 0.078 | 0.284 | 0.161 | 0.206 |
| Team | Run | Genes | GO terms | Exact match | Hierarchical match | ||||
|---|---|---|---|---|---|---|---|---|---|
| P | R | F 1 | hP | hR | hF 1 | ||||
| 183 | 1 | 172 | 860 | 0.117 | 0.157 | 0.134 | 0.322 | 0.356 | 0.338 |
| 183 | 2 | 172 | 1720 | 0.092 | 0.245 | 0.134 | 0.247 | 0.513 | 0.334 |
| 183 | 3 | 172 | 3440 | 0.057 | 0.306 | 0.096 | 0.178 | 0.647 | 0.280 |
| 220 | 1 | 50 | 2639 | 0.018 | 0.075 | 0.029 | 0.064 | 0.190 | 0.096 |
| 220 | 2 | 46 | 1747 | 0.024 | 0.065 | 0.035 | 0.087 | 0.158 | 0.112 |
| 237 | 1 | 23 | 37 | 0.108 | 0.006 | 0.012 | 0.415 | 0.020 | 0.039 |
| 237 | 2 | 96 | 2424 | 0.108 | 0.068 | 0.029 | 0.084 | 0.336 | 0.135 |
| 237 | 3 | 171 | 4631 | 0.037 | 0.264 | 0.064 | 0.150 | 0.588 | 0.240 |
| 238 | 1 | 194 | 1792 | 0.054 | 0.149 | 0.079 | 0.243 | 0.459 | 0.318 |
| 238 | 2 | 194 | 555 | 0.088 | 0.076 | 0.082 | 0.250 | 0.263 | 0.256 |
| 238 | 3 | 194 | 850 | 0.029 | 0.039 | 0.033 | 0.196 | 0.310 | 0.240 |
| 243 | 1 | 109 | 510 | 0.073 | 0.057 | 0.064 | 0.249 | 0.269 | 0.259 |
| 243 | 2 | 104 | 393 | 0.084 | 0.051 | 0.064 | 0.280 | 0.248 | 0.263 |
| 243 | 3 | 144 | 2538 | 0.030 | 0.116 | 0.047 | 0.130 | 0.477 | 0.204 |
| 250 | 1 | 171 | 1389 | 0.052 | 0.112 | 0.071 | 0.174 | 0.328 | 0.227 |
| 250 | 2 | 166 | 1893 | 0.049 | 0.143 | 0.073 | 0.128 | 0.374 | 0.191 |
| 250 | 3 | 132 | 453 | 0.095 | 0.067 | 0.078 | 0.284 | 0.161 | 0.206 |
Discussion
As mentioned earlier, our task is related to a few previous challenge tasks on biomedical text retrieval and semantic indexing. In particular, our task resembles the earlier GO task in BioCreative I ( 22 ). On the other hand, our two subtasks are different from the previous tasks. For the passage retrieval task, we only provide teams with pairs of <gene, document> and asked their systems to return relevant evidence text while <gene, document, GO terms> triples were provided in the earlier task. We provided less input information to teams because we aim to have our tasks resemble real-world GO annotation more closely, where the only input to human curators is the set of documents.
For the GO-term prediction task, we provided teams with the same <gene, document> pairs and asked their systems to return relevant GO terms. In addition to such input pairs, the expected number of GO terms and their associated GO ontologies (Molecular Function, Biological Process, and Cellular Component) returned were also provided in the earlier task. Another difference is that along with each predicted GO term for the given gene in the given document, output of associated evidence text is also required in the earlier task.
The evaluation mechanism also differed in the two challenge events. We provided the reference data before the team submission and preformed standard evaluation. By contrast, in the BioCreative I GO task, no gold-standard evaluation data were provided before the team submission. Instead, expert GO curators were asked to manually judge the team submitted results. Such a posthoc analysis could miss TP results not returned by teams and would not permit evaluation of new systems after the challenge. While there exist other metrics for measuring sentence and semantic similarity ( 31 , 40–42 ), to compare with previous results, we followed the evaluation measures (e.g. precision, recall and F 1 score) in ( 22 ).
Despite these differences, we were intrigued by any potential improvement in the task results due to the advancement of text-mining research in recent years. As the ultimate goal of the task is to find GO terms, the results of Task B are of more interest and significance in this aspect, though evidence sentences are of course important for reaching this goal. By comparing the team results in the two challenge events [ Table 3 above vs. Table 5 in ( 22 )], we can observe a general trend of performance increase on this task over time. For example, the best-performing team in 2005 ( 22 ) was only able to predict 78 TPs (of 1227 in gold standard)—a recall of <7%—while there are several teams in our task who obtained recall values between 10 and 30%. The numbers are even greater when measured by taking account of the hierarchical nature of the Gene Ontology.
Post-challenge analysis: classification of FP sentences
To better understand the types of FP sentences returned by the participating text-mining systems, we asked curators to manually review and classify FP predictions using one or more categories described below. For this analysis, each curator was given three test set papers that they previously annotated. In total, seven curators completed this analysis by assigning 2289 classifications to 2074 sentences.
Sentence classifications
‘ To characterize the functions and interrelationships of CSP41a and CSP41b, T-DNA insertion lines for the genes encoding the two proteins were characterized .’
‘ Molecular studies of the REF-1 family genes hlh-29 and hlh-28 indicate that their gene products are identical, and that loss of hlh-29/hlh-28 activities affects C. elegans embryonic viability, egg-laying, and chemorepulsive behaviors [21] .’
‘ (A) An anti-Aurora A anti-serum recognizes the NH2-terminal recombinant histidine-tagged protein domain used for immunization (left) and the 47-kD endogenous Aurora A protein kinase in Drosophila embryo extracts (right) by Western blotting .’
‘ We found that knockdown of Shank3 specifically impaired mGluR5 signaling at synapses .’
‘ The binding site for AR-C155858 involves TMs 7-10 of MCT1, and probably faces the cytosol .’
‘ These results are in agreement with those obtained for the TIEG3 protein in HeLa and OLI-neu cells [32] and indicate that the Cbt bipartite NLS within the second and third zinc fingers is functional in mammalian cells, suggesting that different nuclear import mechanisms for this protein are being used in Drosophila and mammalian cells .’
‘ Furthermore, in primary macrophages, expression of Fcgr3-rs inhibits Fc receptor-mediated functions, because WKY bone marrow-derived macrophages transduced with Fcgr3-rs had significantly reduced phagocytic activity .’
‘ The atnap null mutant and WT plants are developmentally indistinguishable in terms of bolting and flowering times .’
‘ S5 shows Mad2MDF-2 enrichment on monopolar spindles in the PP1-docking motif mutants .’
‘ Arrows indicate the main CSP41a and CSP41b protein species .’
‘ It is suggested that CSP41 complexes determine the stability of a distinct set of chloroplast transcripts including rRNAs, such that the absence of CSP41b affects both tar-get transcript stability and chloroplast translational activity .’
‘ Thus, mechanistic insight into the reactions that activate checkpoint signaling at the kinetochore and testing the effect of KNL-1 microtubule binding on these reactions as well as elucidating whether KNL-1 mutants participate in parallel to or in the same pathway as dynein in checkpoint silencing are important future goals .’
(1) Experiment was performed —These types of sentences relate that an experiment has been performed but do not describe what the actual result was. Such sentences may or may not contain a GO-related concept.
(2) Previously published result —These sentences refer to experimental findings from papers cited in the test set papers. They often contain a parenthetical reference, or other indication, that the information is from a previously published paper.
(3) Not GOrelated —These sentences describe an aspect of biology that is not amenable to GO curation, i.e. it does not describe a biological process, molecular function or cellular component.
(4) Curator missed —This class of sentences actually represents TP sentences that the curator failed to identify when annotating the test set papers.
(5) Interpretive statements/author speculation —These sentences describe either an author’s broader interpretation of an experimental finding or their speculation on that finding, but do not necessarily provide direct evidence for a GO annotation.
(6) Contiguous sentence —These sentences were selected by curators, but only as part of an annotation that required additional sentences that may or may not be directly adjacent to the annotated sentence. In these cases, curators felt that additional information was needed to completely support the GO annotation.
In this case, information about the assay used to determine ‘these results’ is not available in just this sentence alone.
(7) Sentence was captured —In these cases, the sentence was captured by the curator, but for some, the annotation was for a different gene product than that predicted by the participating teams.
(8) Negative result —These sentences describe an experimental finding, but one for which the result is negative, i.e. the gene product is not involved, and thus the sentence would not be annotated for GO.
(9) Mutant background —These sentences describe an experimental finding that does not reflect the wild-type activity of the gene product. This classification is distinct from sentences that describe mutant phenotypes, which are often used to assign GO Biological Process annotations.
(10) Other —This classification was reserved for sentences that did not readily fit into any of the additional classifications.
(11) Unresolved entity —These sentences mentioned entities, e.g. protein complexes, for which curators were not able to assign a specific ID for annotation.
(12) Future experiments —These sentences describe proposed future experiments.
The results of the classification analysis are presented in Figure 1 . These results indicate that the FP sentences cover a broad range of classifications, but importantly, that only 12% of the FP sentences were classified by curators as completely unrelated to a GO concept. Many of the FP sentences thus contain some element of biology that is relevant to GO annotation, but lack the complete triplet, i.e. an entity, GO term and assay, that is typically required for making a manual experimentally supported GO annotation.
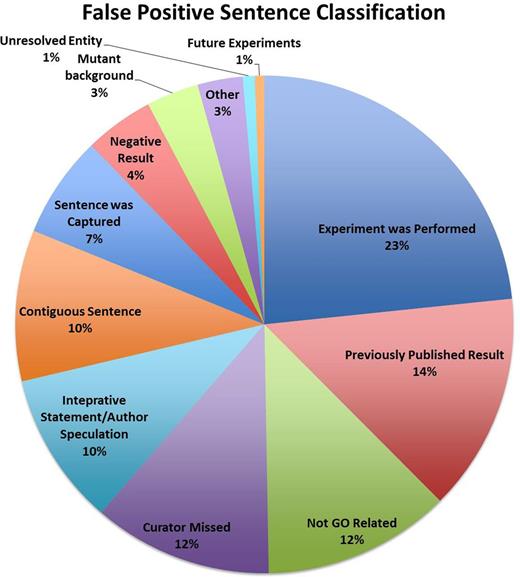
The classification of the FP sentences.
As an additional part of the sentence classification, we also asked curators to indicate which, if any, of the GO triplet was missing from a FP sentence. These analyses indicate some overall trends. For example, many of the sentences that describe previously published results do not indicate the nature of the assay used to determine the experimental findings. In contrast, sentences that describe how or what type of an experiment was performed may include all aspects of the triplet yet lack the actual experimental result that supports an annotation. Likewise, sentences that describe negative results may contain all aspects of the triplet, but the prediction methods failed to discern the lack of association between the gene product and the GO term.
The results of the sentence classification analysis suggest that many of the FP sentences returned by the participating teams have some relevance to GO annotation but either lack one element of the GO annotation triplet or contain all elements of a triplet but fail to correctly discern the actual experimental result. This suggests that further work to refine how evidence sentences are identified and presented to curators may help to improve the utility of text mining for GO annotation. For example, if curation tools can present predicted evidence sentences within the context of the full text of the paper, curators could easily locate those sentences, such as those presented within Results sections, that are most likely to support GO annotations. Additionally, postprocessing of sentences to remove those that contain terms such as ‘not’ or ‘no’ may help to eliminate statements of negative results from consideration.
Further analysis of the content of evidence sentences will hopefully provide valuable feedback to text-mining developers on how to refine their prediction algorithms to improve precision of evidence sentence identification. For example, within a sampling of the largest sentence classification category, ‘Experiment was Performed’, curators marked nearly half of the sentences as containing no GO term. Systematic comparison of these sentences with similarly categorized sentences that did contain a GO term concept may help to improve techniques for GO term recognition.
Additional follow-up analysis may also help annotation groups consider new ways in which to use text-mining results. Text mining for GO annotation might thus expand to include not only predictions for experimentally supported annotations but also predictions for other annotations supported by the text of a paper such as those described from previously published results. GO annotation practice includes an evidence code, Trace-able Author Statement (TAS), that can be used for these types of annotations, so perhaps a new evidence code that indicates a TAS annotation derived from text mining could be developed for such cases.
Individual system descriptions
Each team has agreed to contribute a brief summary of the most notable aspects of their system. In summary, the machine learning approaches performed better than the rule-based approaches in Task A. For example, Team 238 achieved the best performance by using multiple features (bag-of-words, bigram features, section features, topic features, presence of genes) and training a logistic regression model to classify positive vs negative instances of GO evidence sentences.
A variety of methods were attempted for Task B, such as K-nearest-neighbor, pattern matching and information retrieval (IR)-based ranking techniques. Moreover, several participants (Team 183, 238 and 250) used the evidence sentences they retrieved in Task A as input for finding GO terms in Task B. The best performance in Task B was obtained by Team 183’s supervised categorization method, which retrieved most prevalent GO terms among the k most similar instances to the input text in their knowledge base ( 43 ).
Team 183: Julien Gobeill, Patrick Ruch (Task A, Task B)
The BiTeM/SIBtex group participated in the first BioCreative campaign ( 22 ). We then obtained top competitive results, although for all competing systems, performances were far from being useful for the curation community. At this time, we extracted GO terms from full texts with a locally developed dictionary-based classifier ( 44 ). Dictionary-based categorization approaches attempt to exploit lexical similarities between GO terms (descriptions and synonyms) and the input text to be categorized. Such approaches are limited by the complex nature of the GO terms. Identifying GO terms in text is highly challenging, as they often do not appear literally or approximately in text. We have recently reported on GOCat ( 45 , 46 ), our new machine learning GO classifier. GOCat exploits similarities between an input text and already curated instances contained in a knowledge base to infer a functional profile. GO annotations (GOA) and MEDLINE make it possible to exploit a growing amount of almost 100 000 curated abstracts for populating this knowledge base. Moreover, we showed in ( 46 ) that the quality of the GO terms predicted by GOCat continues to improve across the time, thanks to the growing number of high-quality GO terms assignments available in GOA: thus, since 2006, GOCat performances have improved by +50%.
The BioCreative IV Track 4 gave us the opportunity to exploit the GOCat power in a reference challenge. For Task A, we designed a robust state-of-the-art approach, using a naïve Bayes classifier, the official training set and words as features. This approach generally obtained fair results (top performances for high precision systems) and should still benefit from being tuned for this task with the new available benchmark. We also investigated exploiting GeneRIFs for an alternative 40 times bigger training set, but the results were disappointing, probably because of the lack of good-quality negative instances. Then, for Task B, we applied GOCat to the first subtask output and produced three different runs with five, ten or twenty proposed GO terms. These runs outperformed other competing systems both in terms of precision and recall, with performances up to 0.65 for recall with hierarchical metrics. Thanks to BioCreative, we were able to design a complete workflow for curation. Given a gene name and a full text, this system is able to deliver highly relevant GO terms along with a set of evidence sentences. Today, the categorization effectiveness of the tool seems sufficient for being used in real semiautomatic curation workflows, as well as in fully automatic workflow for nonmanually curated biological databases. In particular, GOCat is used to profile PubChem bioassays ( 47 ), and by the COMBREX project to normalize functions described in free text formats ( 48 ).
Team 220: Anh Tuan Luu, Jung-jae Kim (Task B)
Luu and Kim ( 49 ) present a method that is based on the cross products database ( 50 ) and combined with a state-of-the-art statistical method based on the bag of words model. They call the GO concepts that are not defined with cross products, ‘primitive concepts’, where the primitive concepts of a GO term are those that are related to the GO term through cross products possibly in an indirect manner. They assume, like the assumption of bag-of-words approach, that if all or most of the primitive concepts of a GO term appear in a small window of text (e.g. sentence), the GO term is likely to be expressed therein. For each GO term and a text, the method first collects all primitive concepts of the GO term and identifies any expression of a primitive concept in the text. It recognizes as expressions of a primitive concept the words that appear frequently in the documents that are known to express the concept (called domain corpus), but not frequently in a representative subset of all documents (called generic corpus). Given a document δ and a primitive concept γ, if the sum of the relative frequency values of the top-K words of the concept found in the text is larger than a threshold θ, we regard the concept as expressed in the document. Finally, a text is considered to express GO term Γ whose cross products definition has n primitive concepts, if this text expresses at least k primitive concepts among the n concepts, where the value of k is dynamically determined using a sigmoid function, depending on n.
Furthermore, the cross products–based method (called XP method) is incorporated with Gaudan’s method ( 51 ), which shows a better coverage than the XP method, as follows: For each GO term Γ whose cross products definition has n primitive concepts, if the XP method can find evidence to k primitive concepts (as explained above) in the text zone δ, the combined method calculates the sum of the scores from the two methods. If the sum is greater than a threshold, we assume that δ expresses Γ. If a GO term does not have a cross products definition, we only use the score of the Gaudan’s method. In short, we call the combination method is XP-Gaudan method. The experiment results show that the F-measures of the two individual methods are lower than that of the XP-Gaudan method. The recall of the XP-Gaudan method (21%) is close to the sum of the recall values of the two individual methods (26%), which may mean that the two methods target different sets of GO term occurrences. In other words, the XP method is complementary to the Gaudan’s method in detecting GO terms in text documents.
Team 237: Jung-Hsien Chiang, Yu-De Chen, Chia-Jung Yang (Task A, Task B)
We developed two different methods: a sequential pattern mining algorithm and GREPC (Geneontology concept Recognition by Entity, Pattern and Constrain) for the BioCreative GO track to recognize sentences and GO terms.
In our sequential pattern mining algorithm, the highlight of this method is that it can infer GO term and which gene(s) products the GO term belongs to simultaneously. In this method, each of the generated rules has two classes, one for the inferred GO term and another for the GO term to which the gene(s) products belong to. Besides, each of the rules is learned from data without human intervention. The basic idea of the sequential pattern mining algorithm we used was similar to ( 52–55 ). We also used Support and Confidence in association rule learning to measure the rules generated. In this work, the items were terms that appeared in sentences. The different permutations of terms will be considered different patterns because of the spirit of sequential pattern. In the preprocessing, we removed stop words, stemmed the rest and added P.O.S. tagger to each term. Then, we anonymized each of the gene(s) products for generating rules that can be widely used in the situation with different gene product names. For instance, a sentence ‘ In vitro, CSC-1 binds directly to BIR-1 ’ would become ‘ vitro_NN __PROTEIN_0__ bind_VBZ directli_RB __PROTEIN_1__ ’. Both the terms ‘__PROTEIN_0__’ and ‘__PROTEIN_1__’ are anonymized gene(s) products. After preprocessing, we thereafter generated rules from the preprocessed sentences. In the instance we mentioned above, we can generate some rules, e.g. ‘ __PROTEIN_0__ bind_VBZ __PROTEIN_1__ => GO: 0005515, __PROTEIN_0_ ’ and ‘ __PROTEIN_0__ bind_VBZ __PROTEIN_1__ => GO: 0005515, __PROTEIN_1__ ’, where the part before the symbol ‘=>’ is the pattern and after the symbol are the classes. The first class GO: 0005515 is the GO ID. The second class represents the GO term belonging to which anonymized gene(s) products. After all rules have been generated, we used those rules to classify sentences in the testing data.
In the GREPC, we indexed the GO concepts based on three divisions: entity, pattern and constrain. We gathered these kinds of information by text mining inside the GO database ( 56 ). Within that, we reconstructed the semi-structured name and synonyms for a GO concept into a better-structured synonym matrix. With GREPC, we can find GO terms in a sentence with a higher recall without losing much of the precision.
Team 238: Hongfang Liu, Dongqing Zhu (Task A, Task B)
For Task A, the Mayo Clinic system effectively leveraged the learning from positive and unlabeled data approach ( 57 , 58 ) to mitigate the constraint of having limited training data. In addition, the system explored multiple features (e.g. unigrams, bigrams, section type, topic, gene presence, etc.) via a logistic regression model to identify GO evidence sentences. The adopted features in their system brought incremental performance gains, which could be informative to the future design of similar classification systems. Their best performing system achieved 0.27 on exact-F1 and 0.387 for overlap-F1, the highest among all participating systems.
For Task B, the Mayo Clinic team designed two different types of systems: (i) the search-based system predicted GO terms based on existing annotations for GO evidences that are of different textual granularities (i.e. full-text articles, abstracts and sentences) and are obtained by using state-of-the-art IR techniques [i.e. a novel application of the idea of distant supervision in information extraction ( 59 )]; (ii) the similarity-based systems assigned GO terms based on the distance between words in sentences and GO terms/synonyms. While the search-based system significantly outperformed the similarity-based system, a more important finding was that the number and the quality of GO evidence sentences used in the distance supervision largely dictates the effectiveness of distant supervision , meaning a large collection of well-annotated, sentence or paragraph level GO evidences is strongly favored by systems using similar approaches.
Team 243: Ehsan Emadzadeh, Graciela Gonzalez (Task B)
The proposed open-IE approach is based on distributional semantic similarity over the Gene Ontology terms. The technique does not require the annotated data for training, which makes it highly generalizable. Our method finds the related gene functions in a sentence based on semantic similarity of the sentence to GO terms. We use the semantic vectors package ( 60 ) implementation of latent semantic analysis (LSA) ( 61 ) with random indexing ( 62 ) to calculate semantic similarities. GO terms’ semantic vectors are created based on the names of the entries in GO; one semantic vector is created for each term in the ontology. Stop words are removed from GO name, and they are generalized by Porter stemming ( 63 ).
A GO term with the highest semantic similarity to the sentence in GeneGO set will be chosen as the final GO annotation for each gene in the sentence. For example, if a sentence top m (=2) similar GO terms are {g5, g10} and the abstract top n (=5) GO terms are {g4, g8, g5, g2, g9}, then the final predicted GO terms for the sentence related to the gene will be {g5}. m and n are tuning parameters that control the precision and the recall. We found that the first sentences of the paragraphs are the most important sentences in terms of information about gene functions, and including all sentences in a paragraph significantly reduced the precision.
Team 250: Yanpeng Li, Hong Yu (Task A, Task B)
Team 264: Jian-Ming Chen, Hong-Jie Dai (Task A)
To efficiently and precisely retrieve GO information from large amount of biomedical resources, we propose a GO evidence sentence retrieval system conducted via combinatorial applications of semantic class and rule patterns to automatically retrieve GO evidence sentences with specific gene mentions from full-length articles. In our approach, the task is divided into two subtasks: (i) candidate GO sentence retrieval, which selects the candidate GO sentences from a given full text, and (ii) gene entity assignment, which assigns relevant gene mentions to a GO evidence sentence.
In this study, sentences containing gene entities or GO terms are considered as potential evidence sentences. Semantic classes including the adopted and rejected class derived from the training set using semantic-orientation point-wise mutual information (SO-PMI) are used for selecting potential sentences and filtering out FP sentences ( 67 ). To further maximize the performance of GO evidence sentence retrieval, rule patterns generated by domain experts are defined and applied. For example, if a potential sentence matches the rule pattern ‘[GENE].* lead to .*[GO]’, the sentence is selected again as a GO evidence sentence candidate. After generating the sentence candidates, the process of gene entity assignment is performed to identify probable gene mentions contained within each sentence. In our current implementation, a gene is assigned to the sentence S if the gene is mentioned in S . Otherwise, we identify the gene with the maximum occurrence from retrieved sentences in paragraph P in which S belongs, and assign this gene to sentence S . Alternatively, gene with the maximum occurrence from retrieved sentences in article A is verified and assigned to sentence S .
The performance of our GO evidence sentence retrieval system achieves the highest recall of 0.424 and 0.716 in the exact match and relaxed overlap measure, respectively. However, the inadequate F score of our system suggests that the rule patterns used may decrease the system performance. In the future, the conduction of rule selection in rule pattern generation and co-reference resolution in gene entity assignment will be performed to maximize the overall performance.
Conclusions
Based on the comparison of team performance in two BioCreative GO tasks (see details in Discussion), we conclude that the state of the art in automatically mining GO terms from literature has improved over the past decade, and that computer results are getting closer to human performance. But to facilitate real-world GO curation, much progress is still needed to address the remaining technical challenges: First, the number of GO terms (class labels for classification) is extremely large and continues to grow. Second, GO terms (and associated synonyms) are designed for unifying gene function annotations rather than for text mining, and are therefore rarely found verbatim in the article. For example, our analysis shows that only about 1/3 of the annotated GO terms in our corpus can be found using exact matches in their corresponding articles. On the other hand, not every match related to a GO concept is annotated. Instead, only those GO terms that represent experimental findings in a given full-text paper are selected. Hence, automatic methods must be able to filter irrelevant mentions that share names with GO terms (e.g. the GO term ‘growth’ is a common word in articles, but additional contextual information would be required to determine if this relatively high-level term should be used for GO annotation purposes). Although a paper’s title can be very useful in deciding whether it is relevant to a GO concept, any annotations should be attributed to the paper itself rather than its citation. Therefore, excluding the reference section may be a simple suggestion for making these methods more relevant to real-life curation. Finally, human annotation data for building statistical/machine-learning approaches is still lacking. Despite our best efforts, we are only able to include 200 annotated articles in our corpus, which contains evidence text for only 1311 unique gene-GO term combinations.
Our challenge task was inspired and developed in response to the actual needs of GO manual annotation. However, compared with real-world GO annotation, the BioCreative challenge task is simplified in two aspects: (i) gene information is provided to the teams while in reality they are unknown; and (ii) extraction of GO evidence code information is not required for our task while it is an essential part of GO annotation in practice. Further investigation of automatic extraction of gene and evidence code information and their impact in detecting the corresponding GO terms remains as future work.
Acknowledgements
The authors would like to thank Lynette Hirschman, John Wilbur, Cathy Wu, Kevin Cohen, Martin Krallinger and Thomas Wiegers from the BioCreative IV organizing committee for their support, and Judith Blake, Andrew Chatr-aryamontri, Sherri Matis, Fiona McCarthy, Sandra Orchard and Phoebe Roberts from the BioCreative IV User Advisory Group for their helpful discussions.
Funding
This research is supported by NIH Intramural Research Program, National Library of Medicine (Y.M. and Z.L.). The BioCreative IV Workshop is funded by NSF/DBI-0850319. WormBase is funded by National Human Genome Research Institute [U41-HG002223] and the Gene Ontology Consortium by National Human Genome Research Institute (NHGRI) [U41-HG002273]. FlyBase is funded by an NHGRI/NIH grant [U41-HG000739] and the UK Medical Research Council [G1000968]. Team 238 is funded by NSF/ABI-0845523 (H.L. and D.Z.), NIH R01LM009959A1 (H.L. and D.Z.). The SIBtex (Swiss Institute of Bioinformatics) team has been partially supported by the SNF (neXtpresso #153437) and the European Union (Khresmoi #257528).
Conflict of interest . None declared.
References
Author notes
Citation details: Mao,Y., Auken,K.V., Li,D., et al. Overview of the gene ontology task at BioCreative IV. Database (2014) Vol. 2014: article ID bau086; doi:10.1093/database/bau086

{kind=link}