





Abstract
In recent years, thousands of Saccharomyces cerevisiae genomes have been sequenced to varying degrees of completion. The Saccharomyces Genome Database (SGD) has long been the keeper of the original eukaryotic reference genome sequence, which was derived primarily from S. cerevisiae strain S288C. Because new technologies are pushing S. cerevisiae annotation past the limits of any system based exclusively on a single reference sequence, SGD is actively working to expand the original S. cerevisiae systematic reference sequence from a single genome to a multi-genome reference panel. We first commissioned the sequencing of additional genomes and their automated analysis using the AGAPE pipeline. Here we describe our curation strategy to produce manually reviewed high-quality genome annotations in order to elevate 11 of these additional genomes to Reference status.
Database URL : http://www.yeastgenome.org/
Introduction
Recent advances in sequence technology have led to an explosion of available sequence data, and thousands of Saccharomyces cerevisiae genomes have been sequenced to varying degrees of completion in just the past few years ( 1 ). These genomes come from a variety of laboratory and commercial strains, as well as from clinical and environmental isolates. More than 100 of these genomes have been assembled to the level of scaffold or chromosome (see the Genome database at the National Center for Biotechnology Information (NCBI); http://www.ncbi.nlm.nih.gov/genome/genomes/15 ), and have been deposited in the primary sequence databases (GenBank, ENA, DDBJ) that make up the International Nucleotide Sequence Database Collaboration (INSDC; http://www.insdc.org ).
The Saccharomyces Genome Database (SGD; http://www.yeastgenome.org/ ) has long been the keeper of the first eukaryotic genome sequence, which was derived primarily from S. cerevisiae strain S288C and published in 1996 as the output of several years of collaborative work of an international consortium of researchers ( 2 ). This ‘reference genome’ was produced to serve as a single consensus representative S. cerevisiae genome sequence against which all other sequences could be measured. Since that time, SGD has made that original eukaryotic reference sequence and its annotation freely available to researchers around the world, who have thoroughly studied the sequence and put it to use in many great scientific discoveries and breakthroughs ( 3 ).
We are now firmly in the modern era of yeast genomics, in which the rich variety of available genomic sequences has changed the way we study genomes. Comparative genomics is providing clear pictures of the full constituent parts of species’ genomes, which vary not only in nucleotide sequence, but also in gene complements and chromosome architecture. Although much work in yeast genomics has focused on S288C ( 4 ) and its derivatives ( 5 ), a number of different strains are more informative for distinct areas of study [e.g. W303 for aging ( 6 ), SK1 for meiosis and sporulation ( 7 )], and are popularly used in research because their distinct phenotypes.
New technologies have been quickly pushing S. cerevisiae annotation well past the limits of any system based exclusively on a single reference sequence. Therefore, SGD is actively expanding its sequence curation efforts to provide a 12-genome reference panel that allows us to more comprehensively annotate the genetic background studied in experiments for which a strain of known provenance is reported. The 11 additional high-profile yeast genomes were selected because they are widely studied, and have sizable amounts of published experimental and phenotypic data available. When combined with high quality sequence information, these detailed phenotype and functional annotations will allow researchers to fully understand the specific genomic basis of phenotypic variation. Here we present a description of the curation strategy currently in use to expand the original S. cerevisiae systematic reference sequence from a single highly-studied genome to an expertly curated multi-genome reference panel, as shown in Figure 1 .
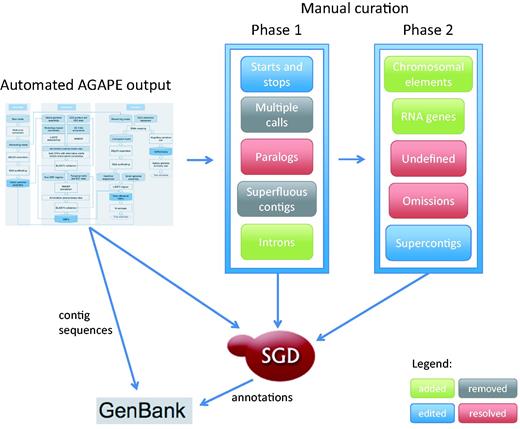
Curation strategy currently in use at SGD to expand the original Saccharomyces cerevisiae systematic reference sequence from a single highly-curated genome to an expertly curated multi-genome reference panel.
Materials and Methods
We recently updated the S288C reference sequence ( 8 ), and then commissioned the sequencing of additional genomes and their automated analysis using the Automated Genome Analysis PipelinE (AGAPE) pipeline (9; NCBI BioProject Accession PRJNA260311). AGAPE accepts raw sequence reads as input, generates de novo assembly scaffolds and contigs, then integrates the steps of open reading frame (ORF) annotation and sequence variation calling. The original AGAPE output is available at http://downloads.yeastgenome.org/published_datasets/Song_2015_PMID_25781462/ . We are now applying the following curation strategy to produce manually reviewed high-quality genome annotations in order to elevate 11 of these additional genomes to Reference status. The 11 strains from which these genome sequences were derived have recently been described elsewhere ( 1 , 9 ). These strains were selected because they have a substantial history of use and experimental results, and also because they are the genomes for which we have the most curated phenotype data and for which we aim to curate specific functional information.
The curation strategy
Step 1: boundary differences. We first identified ORF calls for which the predicted translation start and/or stop differed from the S288C reference, then reviewed them manually and corrected as necessary. These annotation changes and ORF revisions were performed in the same manner that we have used in the past to update the S288C systematic reference sequence, and which we have described previously ( 10 ). For the 11 strains, a total of 1009 such differences were identified ( Table 1 ). Boundary differences fell into three main categories: contig artifacts, intron errors and valid variation. Contig artifacts included ORFs running off the end of contigs and/or ambiguous sequence. In these cases, the pipeline called the closest available in-frame start or stop. Errors due to introns were overwhelmingly in genes that code for ribosomal proteins, especially those in which the leading coding portion is ∼20 nucleotides or less (e.g. RPL2A /YFR031C-A and RPL2B /YIL018W). The remainder of the boundary differences appear valid upon viewing the sequence; we know that some start and stop codons differ between strains (e.g. AQY1 /YPR192W as described in Song et al. , ( 15 )). As further experimental data become available, we will refine these annotations appropriately.
Numbers of automated ORF annotations for 11 different Saccharomyces strains for which the predicted translation start and/or stop generated by the AGAPE sequence analysis pipeline ( 9 ) differed from the S288C reference
| Automated ORFs calls | ORF boundary differences relative to strain S288C | ||||||
|---|---|---|---|---|---|---|---|
| Strain | Provenance | Accession | Start | Stop | Both | Total | |
| CEN.PK | Lab strain | JRIV00000000 | 5379 | 35 | 19 | 39 | 93 |
| D273-10B | Lab strain | JRIY00000000 | 5383 | 37 | 18 | 40 | 95 |
| FL100 | Lab strain | JRIT00000000 | 5366 | 29 | 21 | 34 | 84 |
| JK9-3d | Lab strain | JRIZ00000000 | 5385 | 40 | 11 | 35 | 86 |
| RM11-1a | Vineyard | JRIP00000000 | 5323 | 36 | 17 | 30 | 83 |
| SEY6210 | Lab strain | JRIW00000000 | 5400 | 44 | 23 | 26 | 93 |
| Sigma1278b | Lab strain | JRIQ00000000 | 5358 | 31 | 20 | 28 | 79 |
| SK1 | Lab strain | JRIH00000000 | 5350 | 38 | 22 | 32 | 92 |
| W303 | Lab strain | JRIU00000000 | 5397 | 54 | 24 | 33 | 111 |
| X2180-1A | Lab strain | JRIX00000000 | 5387 | 37 | 24 | 35 | 96 |
| Y55 | Lab strain | JRIF00000000 | 5359 | 39 | 26 | 32 | 97 |
| Total | 420 | 225 | 364 | 1009 | |||
| Automated ORFs calls | ORF boundary differences relative to strain S288C | ||||||
|---|---|---|---|---|---|---|---|
| Strain | Provenance | Accession | Start | Stop | Both | Total | |
| CEN.PK | Lab strain | JRIV00000000 | 5379 | 35 | 19 | 39 | 93 |
| D273-10B | Lab strain | JRIY00000000 | 5383 | 37 | 18 | 40 | 95 |
| FL100 | Lab strain | JRIT00000000 | 5366 | 29 | 21 | 34 | 84 |
| JK9-3d | Lab strain | JRIZ00000000 | 5385 | 40 | 11 | 35 | 86 |
| RM11-1a | Vineyard | JRIP00000000 | 5323 | 36 | 17 | 30 | 83 |
| SEY6210 | Lab strain | JRIW00000000 | 5400 | 44 | 23 | 26 | 93 |
| Sigma1278b | Lab strain | JRIQ00000000 | 5358 | 31 | 20 | 28 | 79 |
| SK1 | Lab strain | JRIH00000000 | 5350 | 38 | 22 | 32 | 92 |
| W303 | Lab strain | JRIU00000000 | 5397 | 54 | 24 | 33 | 111 |
| X2180-1A | Lab strain | JRIX00000000 | 5387 | 37 | 24 | 35 | 96 |
| Y55 | Lab strain | JRIF00000000 | 5359 | 39 | 26 | 32 | 97 |
| Total | 420 | 225 | 364 | 1009 | |||
Numbers of automated ORF annotations for 11 different Saccharomyces strains for which the predicted translation start and/or stop generated by the AGAPE sequence analysis pipeline ( 9 ) differed from the S288C reference
| Automated ORFs calls | ORF boundary differences relative to strain S288C | ||||||
|---|---|---|---|---|---|---|---|
| Strain | Provenance | Accession | Start | Stop | Both | Total | |
| CEN.PK | Lab strain | JRIV00000000 | 5379 | 35 | 19 | 39 | 93 |
| D273-10B | Lab strain | JRIY00000000 | 5383 | 37 | 18 | 40 | 95 |
| FL100 | Lab strain | JRIT00000000 | 5366 | 29 | 21 | 34 | 84 |
| JK9-3d | Lab strain | JRIZ00000000 | 5385 | 40 | 11 | 35 | 86 |
| RM11-1a | Vineyard | JRIP00000000 | 5323 | 36 | 17 | 30 | 83 |
| SEY6210 | Lab strain | JRIW00000000 | 5400 | 44 | 23 | 26 | 93 |
| Sigma1278b | Lab strain | JRIQ00000000 | 5358 | 31 | 20 | 28 | 79 |
| SK1 | Lab strain | JRIH00000000 | 5350 | 38 | 22 | 32 | 92 |
| W303 | Lab strain | JRIU00000000 | 5397 | 54 | 24 | 33 | 111 |
| X2180-1A | Lab strain | JRIX00000000 | 5387 | 37 | 24 | 35 | 96 |
| Y55 | Lab strain | JRIF00000000 | 5359 | 39 | 26 | 32 | 97 |
| Total | 420 | 225 | 364 | 1009 | |||
| Automated ORFs calls | ORF boundary differences relative to strain S288C | ||||||
|---|---|---|---|---|---|---|---|
| Strain | Provenance | Accession | Start | Stop | Both | Total | |
| CEN.PK | Lab strain | JRIV00000000 | 5379 | 35 | 19 | 39 | 93 |
| D273-10B | Lab strain | JRIY00000000 | 5383 | 37 | 18 | 40 | 95 |
| FL100 | Lab strain | JRIT00000000 | 5366 | 29 | 21 | 34 | 84 |
| JK9-3d | Lab strain | JRIZ00000000 | 5385 | 40 | 11 | 35 | 86 |
| RM11-1a | Vineyard | JRIP00000000 | 5323 | 36 | 17 | 30 | 83 |
| SEY6210 | Lab strain | JRIW00000000 | 5400 | 44 | 23 | 26 | 93 |
| Sigma1278b | Lab strain | JRIQ00000000 | 5358 | 31 | 20 | 28 | 79 |
| SK1 | Lab strain | JRIH00000000 | 5350 | 38 | 22 | 32 | 92 |
| W303 | Lab strain | JRIU00000000 | 5397 | 54 | 24 | 33 | 111 |
| X2180-1A | Lab strain | JRIX00000000 | 5387 | 37 | 24 | 35 | 96 |
| Y55 | Lab strain | JRIF00000000 | 5359 | 39 | 26 | 32 | 97 |
| Total | 420 | 225 | 364 | 1009 | |||
Step 2: multiple calls and paralogs. We examined ORFs that had been called on more than one contig ( Table 2 ). Through manual review we selected the best call for each ORF in this set based on contig size and sequence quality, then discarded the unneeded duplicate, triplicate or quadruplicate annotations. We also looked at calls for ORFs that are members of paralogous pairs, which accounted for many of the duplicates. The S. cerevisiae genome contains over 500 sets of paralogs ( 11–13 ). Often the automated pipeline annotated both paralogous ORFs as the same paralog. These annotation errors are easily identifiable based on up- and downstream neighbors. The majority of ORFs identified erroneously during the automated annotation were those coding for ribosomal proteins (e.g. RPL33A /YPL143W and RPL33B /YOR234C), chaperones (e.g. SSB1 /YDL229W and SSB2 /YNL209W) and transporters (e.g. GEX1 /YCL073C and GEX2 /YKR106W; VBA3 /YCL069W and VBA5 /YKR105C).
Numbers of ORFs in 11 different S. cerevisiae strains that the AGAPE sequence analysis pipeline ( 9 ) called on more than one contig
| ORFs called on multiple contigs | ||||
|---|---|---|---|---|
| Strain | Two contigs | Three contigs | Four contigs | Total |
| CEN.PK | 15 | 3 | 4 | 22 |
| D273-10B | 14 | 6 | 3 | 23 |
| FL100 | 12 | 4 | 3 | 19 |
| JK9-3d | 8 | 3 | 3 | 14 |
| RM11-1a | 12 | 3 | 3 | 18 |
| SEY6210 | 19 | 3 | 4 | 26 |
| Sigma1278b | 14 | 2 | 3 | 19 |
| SK1 | 15 | 2 | 5 | 22 |
| W303 | 36 | 1 | 2 | 39 |
| X2180-1A | 15 | 2 | 3 | 20 |
| Y55 | 25 | 2 | 5 | 32 |
| Total | 185 | 31 | 38 | 254 |
| ORFs called on multiple contigs | ||||
|---|---|---|---|---|
| Strain | Two contigs | Three contigs | Four contigs | Total |
| CEN.PK | 15 | 3 | 4 | 22 |
| D273-10B | 14 | 6 | 3 | 23 |
| FL100 | 12 | 4 | 3 | 19 |
| JK9-3d | 8 | 3 | 3 | 14 |
| RM11-1a | 12 | 3 | 3 | 18 |
| SEY6210 | 19 | 3 | 4 | 26 |
| Sigma1278b | 14 | 2 | 3 | 19 |
| SK1 | 15 | 2 | 5 | 22 |
| W303 | 36 | 1 | 2 | 39 |
| X2180-1A | 15 | 2 | 3 | 20 |
| Y55 | 25 | 2 | 5 | 32 |
| Total | 185 | 31 | 38 | 254 |
Numbers of ORFs in 11 different S. cerevisiae strains that the AGAPE sequence analysis pipeline ( 9 ) called on more than one contig
| ORFs called on multiple contigs | ||||
|---|---|---|---|---|
| Strain | Two contigs | Three contigs | Four contigs | Total |
| CEN.PK | 15 | 3 | 4 | 22 |
| D273-10B | 14 | 6 | 3 | 23 |
| FL100 | 12 | 4 | 3 | 19 |
| JK9-3d | 8 | 3 | 3 | 14 |
| RM11-1a | 12 | 3 | 3 | 18 |
| SEY6210 | 19 | 3 | 4 | 26 |
| Sigma1278b | 14 | 2 | 3 | 19 |
| SK1 | 15 | 2 | 5 | 22 |
| W303 | 36 | 1 | 2 | 39 |
| X2180-1A | 15 | 2 | 3 | 20 |
| Y55 | 25 | 2 | 5 | 32 |
| Total | 185 | 31 | 38 | 254 |
| ORFs called on multiple contigs | ||||
|---|---|---|---|---|
| Strain | Two contigs | Three contigs | Four contigs | Total |
| CEN.PK | 15 | 3 | 4 | 22 |
| D273-10B | 14 | 6 | 3 | 23 |
| FL100 | 12 | 4 | 3 | 19 |
| JK9-3d | 8 | 3 | 3 | 14 |
| RM11-1a | 12 | 3 | 3 | 18 |
| SEY6210 | 19 | 3 | 4 | 26 |
| Sigma1278b | 14 | 2 | 3 | 19 |
| SK1 | 15 | 2 | 5 | 22 |
| W303 | 36 | 1 | 2 | 39 |
| X2180-1A | 15 | 2 | 3 | 20 |
| Y55 | 25 | 2 | 5 | 32 |
| Total | 185 | 31 | 38 | 254 |
Step 3: superfluous contigs. Due to the nature of high-throughput sequencing, a number of redundant contigs were generated for each of the 11 genomes. Because they unnecessarily complicate the genome annotation, they have been removed from the sequence files, reducing the number of contigs by more than half ( Table 3 ). These contigs included those on which no genes were called, most often due to short overall length (e.g. JRIP01000320.1 from strain RM11-1A) or ambiguous sequence (e.g., JRIT01000109.1 in strain FL100).
Numbers of contigs for 11 different S. cerevisiae strains in the original automated output from the AGAPE sequence analysis pipeline ( 9 ) and in the curated contig set after manual review
| Contig set | ||
|---|---|---|
| Strain | Original | Curated |
| CEN.PK | 389 | 189 |
| D273-10B | 403 | 203 |
| FL100 | 402 | 174 |
| JK9-3d | 431 | 197 |
| RM11-1a | 325 | 169 |
| SEY6210 | 366 | 183 |
| Sigma1278b | 451 | 206 |
| SK1 | 389 | 214 |
| W303 | 415 | 236 |
| X2180-1A | 409 | 212 |
| Y55 | 413 | 198 |
| Total | 4393 | 2181 |
| Contig set | ||
|---|---|---|
| Strain | Original | Curated |
| CEN.PK | 389 | 189 |
| D273-10B | 403 | 203 |
| FL100 | 402 | 174 |
| JK9-3d | 431 | 197 |
| RM11-1a | 325 | 169 |
| SEY6210 | 366 | 183 |
| Sigma1278b | 451 | 206 |
| SK1 | 389 | 214 |
| W303 | 415 | 236 |
| X2180-1A | 409 | 212 |
| Y55 | 413 | 198 |
| Total | 4393 | 2181 |
Numbers of contigs for 11 different S. cerevisiae strains in the original automated output from the AGAPE sequence analysis pipeline ( 9 ) and in the curated contig set after manual review
| Contig set | ||
|---|---|---|
| Strain | Original | Curated |
| CEN.PK | 389 | 189 |
| D273-10B | 403 | 203 |
| FL100 | 402 | 174 |
| JK9-3d | 431 | 197 |
| RM11-1a | 325 | 169 |
| SEY6210 | 366 | 183 |
| Sigma1278b | 451 | 206 |
| SK1 | 389 | 214 |
| W303 | 415 | 236 |
| X2180-1A | 409 | 212 |
| Y55 | 413 | 198 |
| Total | 4393 | 2181 |
| Contig set | ||
|---|---|---|
| Strain | Original | Curated |
| CEN.PK | 389 | 189 |
| D273-10B | 403 | 203 |
| FL100 | 402 | 174 |
| JK9-3d | 431 | 197 |
| RM11-1a | 325 | 169 |
| SEY6210 | 366 | 183 |
| Sigma1278b | 451 | 206 |
| SK1 | 389 | 214 |
| W303 | 415 | 236 |
| X2180-1A | 409 | 212 |
| Y55 | 413 | 198 |
| Total | 4393 | 2181 |
Step 4: RNAs and chromosomal elements. One limitation of the automated AGAPE annotation is that it focuses solely on protein-coding genes. Many other types of genes and chromosomal elements can be identified through BLAST, then verified and refined through manual curation. We have identified all 16 centromeres in all 11 strains, and are currently working to identify the many RNA genes (e.g. snRNAs, snoRNAs, tRNAs) and replication origins. This curation work will be ongoing over the coming months.
Step 5: the undefined. We are currently working to identify the 1699 ORFs that were marked ‘unidentifiable’ through the AGAPE pipeline ( Table 4 ). Some of these are indeed identifiable but need annotation updates to account for missed introns and coding segments (e.g. TDA5 /YLR426W). We expect that the majority of the undefined are actually known ORFs that will ultimately be deleted from the annotation due to ambiguous sequence (e.g. ALD2 /YMR170C) or because they are truncated at the ends of contigs, often in most or all 11 strains (e.g. SSA1 /YAL005C and COS7 /YDL248W). At least some of the undefined represent bona fide novel ORFs (e.g. YER065W-A in strain JK9-3d).
Numbers of ORFs 11 different S. cerevisiae strains that were marked as ‘unidentifiable’ in the original automated output from the AGAPE sequence analysis pipeline ( 9 ). These ORFs are currently undergoing manual review
| Strain | Undefined ORFs |
|---|---|
| CEN.PK | 169 |
| D273-10B | 254 |
| FL100 | 128 |
| JK9-3d | 121 |
| RM11-1a | 344 |
| SEY6210 | 69 |
| Sigma1278b | 106 |
| SK1 | 124 |
| W303 | 158 |
| X2180-1A | 78 |
| Y55 | 148 |
| Total | 1699 |
| Strain | Undefined ORFs |
|---|---|
| CEN.PK | 169 |
| D273-10B | 254 |
| FL100 | 128 |
| JK9-3d | 121 |
| RM11-1a | 344 |
| SEY6210 | 69 |
| Sigma1278b | 106 |
| SK1 | 124 |
| W303 | 158 |
| X2180-1A | 78 |
| Y55 | 148 |
| Total | 1699 |
Numbers of ORFs 11 different S. cerevisiae strains that were marked as ‘unidentifiable’ in the original automated output from the AGAPE sequence analysis pipeline ( 9 ). These ORFs are currently undergoing manual review
| Strain | Undefined ORFs |
|---|---|
| CEN.PK | 169 |
| D273-10B | 254 |
| FL100 | 128 |
| JK9-3d | 121 |
| RM11-1a | 344 |
| SEY6210 | 69 |
| Sigma1278b | 106 |
| SK1 | 124 |
| W303 | 158 |
| X2180-1A | 78 |
| Y55 | 148 |
| Total | 1699 |
| Strain | Undefined ORFs |
|---|---|
| CEN.PK | 169 |
| D273-10B | 254 |
| FL100 | 128 |
| JK9-3d | 121 |
| RM11-1a | 344 |
| SEY6210 | 69 |
| Sigma1278b | 106 |
| SK1 | 124 |
| W303 | 158 |
| X2180-1A | 78 |
| Y55 | 148 |
| Total | 1699 |
Steps 6 and 7: omissions and supercontigs. Curation work in the coming year will focus on annotating essential, conserved protein-coding genes that we expect are present in the other genomes but escaped automated annotation undetected. We will also assess several contig pairs for the possibility of combining them into supercontigs (e.g. contigs JRIU01000255.1 and JRIU01000122.1 from strain W303).
Future Directions
The expansion in SGD from a single reference genome to a multi-genome reference panel furthers yeast genomics research by providing easy access to alternative alleles and sequence variants ( 14 ). The automated annotations for the additional genomes, as produced by the AGAPE pipeline, have already been incorporated into SGD sequence and alignment pages, analysis tools such as BLAST, PatMatch, and the Variant Viewer, and are also available for download ( Table 5 ). The application of manual curation to automated output improves the quality and increases the depth and granularity of genome annotation. Curated sequence files and annotation will be incorporated into SGD and also submitted to NCBI’s GenBank primary sequence repository within the coming year. As further research by the scientific community provides updated information, we will incorporate improved annotations into future genome releases. Consideration will also be given in the future to the possibility of expanding the reference panel to accommodate emerging or underserved areas of study, as part of our continuing efforts to educate students, enable bench researchers and facilitate scientific discovery.
Additional S. cerevisiae strain genome sequences are already available throughout SGD
| Location | URL |
|---|---|
| Alignment pages | http://www.yeastgenome.org/cgi-bin/FUNGI/alignment.pl?locus=sal1 |
| BLAST | http://www.yeastgenome.org/blast-sgd |
| Downloads | http://www.yeastgenome.org/download-data/sequence |
| Sequence pages | http://www.yeastgenome.org/locus/sal1/sequence |
| Pattern matching | http://www.yeastgenome.org/cgi-bin/PATMATCH/nph-patmatch |
| Variant viewer | http://www.yeastgenome.org/variant-viewer |
| Location | URL |
|---|---|
| Alignment pages | http://www.yeastgenome.org/cgi-bin/FUNGI/alignment.pl?locus=sal1 |
| BLAST | http://www.yeastgenome.org/blast-sgd |
| Downloads | http://www.yeastgenome.org/download-data/sequence |
| Sequence pages | http://www.yeastgenome.org/locus/sal1/sequence |
| Pattern matching | http://www.yeastgenome.org/cgi-bin/PATMATCH/nph-patmatch |
| Variant viewer | http://www.yeastgenome.org/variant-viewer |
Additional S. cerevisiae strain genome sequences are already available throughout SGD
| Location | URL |
|---|---|
| Alignment pages | http://www.yeastgenome.org/cgi-bin/FUNGI/alignment.pl?locus=sal1 |
| BLAST | http://www.yeastgenome.org/blast-sgd |
| Downloads | http://www.yeastgenome.org/download-data/sequence |
| Sequence pages | http://www.yeastgenome.org/locus/sal1/sequence |
| Pattern matching | http://www.yeastgenome.org/cgi-bin/PATMATCH/nph-patmatch |
| Variant viewer | http://www.yeastgenome.org/variant-viewer |
| Location | URL |
|---|---|
| Alignment pages | http://www.yeastgenome.org/cgi-bin/FUNGI/alignment.pl?locus=sal1 |
| BLAST | http://www.yeastgenome.org/blast-sgd |
| Downloads | http://www.yeastgenome.org/download-data/sequence |
| Sequence pages | http://www.yeastgenome.org/locus/sal1/sequence |
| Pattern matching | http://www.yeastgenome.org/cgi-bin/PATMATCH/nph-patmatch |
| Variant viewer | http://www.yeastgenome.org/variant-viewer |
Funding
This work was supported by a U41 grant from the National Human Genome Research Institute at the US National Institutes of Health (HG001315) to the Saccharomyces Genome Database project. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Human Genome Research Institute or the National Institutes of Health.
Conflict of interest . None declared.
References
Author notes
Citation details: Engel,S.R., Weng,S., Binkley,G., et al. From one to many: expanding the Saccharomyces cerevisiae reference genome panel. Database (2016) Vol. 2016: article ID baw020; doi:10.1093/database/baw020

{kind=link}