





Abstract
The significant amount of medicinal chemistry information contained in patents makes them an attractive target for text mining. In this manuscript, we describe systems for named entity recognition (NER) of chemicals and genes/proteins in patents, using the CEMP (for chemicals) and GPRO (for genes/proteins) corpora provided by the CHEMDNER task at BioCreative V. Our chemical NER system is an ensemble of five open systems, including both versions of tmChem, our previous work on chemical NER. Their output is combined using a machine learning classification approach. Our chemical NER system obtained 0.8752 precision and 0.9129 recall, for 0.8937 f-score on the CEMP task. Our gene/protein NER system is an extension of our previous work for gene and protein NER, GNormPlus. This system obtained a performance of 0.8143 precision and 0.8141 recall, for 0.8137 f-score on the GPRO task. Both systems achieved the highest performance in their respective tasks at BioCreative V. We conclude that an ensemble of independently-created open systems is sufficiently diverse to significantly improve performance over any individual system, even when they use a similar approach.
Database URL: http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/tmTools/ .
Introduction
While publications such as those found in the biomedical literature contain a significant amount of useful chemical information ( 1 ), much of the useful information on medicinal chemistry is found in less formal documents, such as patents. The CHEMDNER task at BioCreative V, a major challenge event in biomedical natural language processing, addressed the extraction of chemical and gene/protein entities from medicinal chemistry patents ( 2 ). A sentence with example annotations of both a chemical and a protein is shown in Figure 1 . NCBI participated in both the CEMP [chemical named entity recognition (NER)] and GPRO (gene and protein related object identification) subtasks. We addressed the CEMP subtask with an ensemble system combining the results of 10 models from five open NER systems for chemical NER. We addressed the GPRO subtask by adapting the open source GNormPlus system ( 3 ).

An example sentence from the CHEMDNER training corpus with chemicals and gene/proteins in Patent ID: CA2119782C.
The corpus for the BioCreative V CHEMDNER task was generated using the annotation guidelines from the BioCreative IV CHEMDNER task, with slight differences to adapt the guidelines to patents. These differences increase the focus on capturing the broad chemical terminology rather than specifically on mentions that can be converted to a chemical structure. As in the BioCreative IV CHEMDNER task, seven types of chemical (CEMP) mentions are highlighted: systematic, identifiers, formula, trivial, abbreviation, family and multiple. Several high performance open source chemical recognition systems were developed during the CHEMDNER task, in which the best performance was over 87% f-score ( 1 ). A description of the CHEMDNER patent corpus and its CEMP mentions can be found in Table 1 .
Description of the training, development and test sets for the BioCreative V CHEMDNER task, including mentions for the chemical named entity recognition (CEMP) and gene and protein related object identification (GPRO) subtasks
| Count description | Training set | Development set | Test set |
|---|---|---|---|
| Patent abstracts | 7000 | 7000 | 7000 |
| CEMP mentions | 33543 | 32142 | 33949 |
| GPRO mentions | 6876 | 6263 | 7093 |
| Type 1 only | 4396 | 3934 | 4093 |
| Count description | Training set | Development set | Test set |
|---|---|---|---|
| Patent abstracts | 7000 | 7000 | 7000 |
| CEMP mentions | 33543 | 32142 | 33949 |
| GPRO mentions | 6876 | 6263 | 7093 |
| Type 1 only | 4396 | 3934 | 4093 |
Description of the training, development and test sets for the BioCreative V CHEMDNER task, including mentions for the chemical named entity recognition (CEMP) and gene and protein related object identification (GPRO) subtasks
| Count description | Training set | Development set | Test set |
|---|---|---|---|
| Patent abstracts | 7000 | 7000 | 7000 |
| CEMP mentions | 33543 | 32142 | 33949 |
| GPRO mentions | 6876 | 6263 | 7093 |
| Type 1 only | 4396 | 3934 | 4093 |
| Count description | Training set | Development set | Test set |
|---|---|---|---|
| Patent abstracts | 7000 | 7000 | 7000 |
| CEMP mentions | 33543 | 32142 | 33949 |
| GPRO mentions | 6876 | 6263 | 7093 |
| Type 1 only | 4396 | 3934 | 4093 |
Recognition of mentions of genes/proteins, diseases and related objects in biomedical literature has been well studied in the past decade ( 4–7 ), including through multiple community-wide text-mining challenges ( 8 ). The best performance is over 80% f-score for both recognizing gene/protein mentions and normalizing the mentions to specific gene/protein identifiers within a controlled vocabulary (e.g. NCBI Gene or UniProt). Unlike previous gene recognition/normalization tasks ( 9–13 ), the BioCreative V GPRO subtask attempts to identify genes and proteins in patent texts. Under the criteria of the task, only gene/protein name mentions (named type 1 in this task), which can be normalized to a database (e.g. UniProt and NCBI Gene) identifier are evaluated. Other related entities, such as gene/protein families, domains, sequences, motifs and DNA/RNA (named type 2 in this task) are not considered in the evaluation. The main challenge in this task therefore becomes how to recognize the difference between genes and proteins that can be normalized and other objects. A description of the GPRO mentions in the CHEMDNER patent corpus can also be found in Table 1 .
We address both the CEMP and GPRO tasks with an ensemble approach, combining the results of several models to improve performance. Variations of ensembles have been used in machine learning for many years; one well-known method—bagging—uses bootstrap samples of the training data to train multiple models, then averages their predictions ( 14 ). Attempts to address NER with ensemble systems vary greatly in the composition of the ensemble and the methodology used for combining the predictions. At the gene/protein NER task at the first BioCreative challenge, one participant combined a support vector machine and two hidden markov models using majority vote ( 15 ). Other gene/protein named entity recognizers have used conditional random fields models exclusively by training the same model using different tagging directions, then combining using their tagging probabilities ( 16 ) or by training the same model on corpus-specific alternate annotations, then combining with rule-based strategies ( 17 ). Our ensemble approach is similar to methods combining team results ( 13 , 18 ).
Ensemble techniques have been found to be particularly effective in noisy domains. For example, a participant in the 2010 i2/b2 NLP Challenge combined two dictionary systems and five machine learning systems using majority vote ( 19 ). These results suggest that ensemble methods should be useful in patents, which are also somewhat noisy. Since system diversity is typically considered to be important for an ensemble ( 19 ), however, it is still somewhat unclear whether a combination of conditional random field systems with broadly similar approaches that have not been specifically engineered to be different will produce increased performance.
CEMP task methods
We addressed the CEMP subtask using an ensemble system that combines the results from five individual systems, trained with different data to create a total of ten models. An overview of the system architecture is depicted in Figure 2 . Each of the individual systems included are machine learning systems based on conditional random fields and employ a rich feature approach. The systems used are tmChem Model 1 and tmChem Model 2 ( 4 ), becas[chemical] ( 20 ), the Wuhan university CHEMDNER tagger ( 21 ) and banner-chemdner ( 22 ). All systems are retrainable, with the exception of becas[chemical], which is only available as a web service.
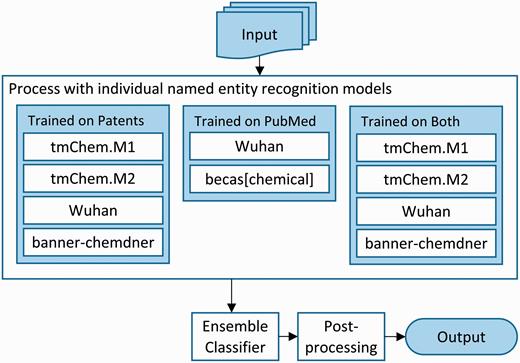
Architecture of the ensemble chemical named entity recognition system for the CEMP task.
We trained the constituent systems using combinations of two corpora. First, we used the training and development sets of annotated patents provided by the organizers. We pooled these sets and randomly split them into three sets: the training set, containing 12 000 articles, and two evaluation sets containing 1000 articles each. Second, we also used the full corpus of PubMed abstracts from the BioCreative IV CHEMDNER task as a training corpus ( 23 ). We created ten separate models: four models using only the patents training data, two models using only the PubMed data and four models using both, as shown in Figure 2 .
We combined the results of the ten models using a machine learning classification approach. Each mention returned by at least one model was represented as an instance to be classified. We used 10 binary features—one feature per model—with the value of the feature reflecting whether the respective model returned the mention. We tested several classifiers, including majority vote, logistic regression and support vector machines (both of which learn weights for each feature) and random forests (which considers feature interactions). Majority vote performed much better than random forests, but we selected logistic regression and support vector machines because they provided the highest performance. Our implementation used Weka ( 24 ) and libsvm ( 25 ), with the default parameters for each classifier (a grid search found no configuration with higher performance).
We handle overlapping mentions by selecting the mention with the highest classification score, in case of a tie we use the longer mention. This changes the precision/recall balance, which we address by determining the optimal classification score threshold for each classifier on the two evaluation sets, we use their average for the final ensemble.
We created two versions of the ensemble with the intention of maximizing recall. The first (‘high recall’) omits the thresholding step, returning all mentions found after handling overlaps. The second (‘higher recall’) also omits the thresholding step and adds an additional post-processing step. This step considers each text that was marked as a mention by the ensemble and then searches the document for other instances of that text. If another instance is found, then it is also added as a chemical mention, unless it was already present.
GPRO task methods
We addressed the GPRO task by adapting GNormPlus ( 3 ), our previous work on gene/protein name recognition and normalization. GNormPlus is a conditional random fields (CRF) ( 26 ) based method which can recognize gene/protein, family and domain mentions, and also determine their respective identifiers in NCBI Gene. By default, GNormPlus is trained using the refactored corpus of BioCreative II Gene Normalization task ( 11 ).
For the GPRO task, we used the BIEO (B: begin, I: inside, E: end and O: outside) labeling model and a CRF order of 2. More specifically, we created five individual models (M1-M5) based on different training data and features, as shown in Table 2 . We first separated all gene/protein-related annotations into two distinct types: mentions that can be normalized to a database record (type 1) and mentions that cannot (type 2). Next, our five models were designed as follows: In model 1, both types of mentions were used and were treated the same. In model 2, both types were used but treated as two separate classes. In model 3, the type 2 mentions were ignored and the model was trained with only the type 1 mentions. Models 4 and 5 resembled models 1 and 3 respectively, but also used the recognition result of the default GNormPlus system as an additional feature. The other features used in the five models were directly adapted from GNormPlus, including linguistic features, character calculation, semantic type and contextual words.
Detailed description of the five models for GPRO task
| Model | Details |
|---|---|
| M1 | Use both Type 1 and 2 mentions, treat them as a single class |
| M2 | Use both Type 1 and 2 mentions, but treat as two distinct classes |
| M3 | Use only Type 1 mentions, ignore Type 2 mentions |
| M4 | Like M1, but with the additional GNormPlus feature |
| M5 | Like M3, but with the additional GNormPlus feature |
| Model | Details |
|---|---|
| M1 | Use both Type 1 and 2 mentions, treat them as a single class |
| M2 | Use both Type 1 and 2 mentions, but treat as two distinct classes |
| M3 | Use only Type 1 mentions, ignore Type 2 mentions |
| M4 | Like M1, but with the additional GNormPlus feature |
| M5 | Like M3, but with the additional GNormPlus feature |
The additional GNormPlus feature refers to the results of the default GNormPlus model, trained on PubMed abstracts.
Detailed description of the five models for GPRO task
| Model | Details |
|---|---|
| M1 | Use both Type 1 and 2 mentions, treat them as a single class |
| M2 | Use both Type 1 and 2 mentions, but treat as two distinct classes |
| M3 | Use only Type 1 mentions, ignore Type 2 mentions |
| M4 | Like M1, but with the additional GNormPlus feature |
| M5 | Like M3, but with the additional GNormPlus feature |
| Model | Details |
|---|---|
| M1 | Use both Type 1 and 2 mentions, treat them as a single class |
| M2 | Use both Type 1 and 2 mentions, but treat as two distinct classes |
| M3 | Use only Type 1 mentions, ignore Type 2 mentions |
| M4 | Like M1, but with the additional GNormPlus feature |
| M5 | Like M3, but with the additional GNormPlus feature |
The additional GNormPlus feature refers to the results of the default GNormPlus model, trained on PubMed abstracts.
As in GNormPlus, we employed several post-processing steps: including enforcing tagging consistency and abbreviation resolution. In addition, we performed filtering, especially for the false positive predictions in two major types: ‘gene/protein family name’ and ‘not a gene/protein mention.’ We filtered these using a maximum entropy classifier trained with three types of features: the five tokens surrounding the span, whether the span can be found in NCBI Gene, UniProt or the list of type 1 mentions in the training and development sets, and morphological features: The number of uppercases, lowercases, digits, tokens, and binary features of common gene/protein (e.g. ‘alpha’) or family (e.g. ‘proteins’) suffixes.
We also filtered composite mentions (‘MULTIPLE’ type) by applying our previous study SimConcept ( 27 ) to recognize these mentions rather than simplify them. The mentions are recognized as mention with coordination ellipsis or range mention are definitely type 2 and should be removed from our output result. The individual mentions (e.g. ‘hdac2 and/or hdac3’) should be separated to multiple spans (‘hdac2’ and ‘hdac3’). However it is difficult to determine whether overlapping abbreviation pair mentions (e.g. ‘NMDA (N-methyl-D-aspartate) receptor’) belong to type 1 or 2. Thus, our system looks at the two individual mentions (‘NMDA receptor’ and ‘N-methyl-D-aspartate receptor’), which are identified by SimConcept. These are ignored if there is a type 2 mention in training corpus with the same text, otherwise they are kept (e.g. ‘tumor necrosis factor (TNF)-a’).
We observed that in the training corpus, some chemical identifiers are recognized as gene/proteins (e.g. ‘KRP-101’ in patent ID: WO2006090756A1). Thus, we applied the lexicon of chemical identifiers which was used in tmChem. This lexicon is collected from the CTD database ( http://ctdbase.org/ ) by extracting the chemical names consisting of 2–5 letters, followed by at least two digits.
Taken together, these post-processing steps improve the f-score by 3–5% on the development set. For the final task submissions, we created two variants that used majority voting to aggregate the results of multiple individual models.
CEMP task results
We evaluated our ensemble and the individual models created for the CEMP task in terms of precision, recall and f-score, requiring the predicted span to match the span annotated to consider it a true positive. We report the performance of both versions of tmChem on our two evaluation sets for the CEMP task in Table 3 . We found that applying models trained on PubMed abstracts to the patent corpus reduced performance as much as 20% (data not shown). Performance improved considerably when the systems were retrained on the patent set, as would be expected. Less expected, however, is that training with a combination of the PubMed and patent sets consistently resulted in a slightly higher net f-score, due to higher recall.
Results for tmChem model 1 and model 2 on the CEMP task in two training configurations
| System | Training | Precision | Recall | F-score |
|---|---|---|---|---|
| tmChem.M1 | Patent | 0.8819 | 0.8088 | 0.8437 |
| tmChem.M2 | Patent | 0.8721 | 0.7953 | 0.8319 |
| tmChem.M1 | Both | 0.8741 | 0.8232 | 0.8479 |
| tmChem.M2 | Both | 0.8711 | 0.8159 | 0.8426 |
| System | Training | Precision | Recall | F-score |
|---|---|---|---|---|
| tmChem.M1 | Patent | 0.8819 | 0.8088 | 0.8437 |
| tmChem.M2 | Patent | 0.8721 | 0.7953 | 0.8319 |
| tmChem.M1 | Both | 0.8741 | 0.8232 | 0.8479 |
| tmChem.M2 | Both | 0.8711 | 0.8159 | 0.8426 |
Each measure is averaged between the two evaluation sets. The highest value is shown in bold.
Results for tmChem model 1 and model 2 on the CEMP task in two training configurations
| System | Training | Precision | Recall | F-score |
|---|---|---|---|---|
| tmChem.M1 | Patent | 0.8819 | 0.8088 | 0.8437 |
| tmChem.M2 | Patent | 0.8721 | 0.7953 | 0.8319 |
| tmChem.M1 | Both | 0.8741 | 0.8232 | 0.8479 |
| tmChem.M2 | Both | 0.8711 | 0.8159 | 0.8426 |
| System | Training | Precision | Recall | F-score |
|---|---|---|---|---|
| tmChem.M1 | Patent | 0.8819 | 0.8088 | 0.8437 |
| tmChem.M2 | Patent | 0.8721 | 0.7953 | 0.8319 |
| tmChem.M1 | Both | 0.8741 | 0.8232 | 0.8479 |
| tmChem.M2 | Both | 0.8711 | 0.8159 | 0.8426 |
Each measure is averaged between the two evaluation sets. The highest value is shown in bold.
We evaluated five versions of the ensemble, as shown in Table 4 . The first three versions differed only in which classifier was used for the ensemble: logistic regression, libsvm and support vector machines with the modified Huber loss. The last two versions were intended to maximize recall, as described in the CEMP Task Methods section; both used logistic regression as the base classifier. To evaluate the ensemble internally, we applied cross-validation between the two evaluation sets and averaged the results. To apply the ensemble to the test set, we included the evaluation sets in the training data for the ensemble classifier, but we did not retrain the individual models.
Results for our ensemble systems on the CEMP task as measured by precision (P), recall (R) and f-score (F)
| System | Evaluation sets | Test set | ||||
|---|---|---|---|---|---|---|
| Precision | Recall | F-score | Precision | Recall | F-score | |
| Logistic | 0.8867 | 0.8979 | 0.8923 | 0.8752 | 0.9129 | 0.8937 |
| Huber SVM | 0.9091 | 0.8626 | 0.8853 | 0.8908 | 0.8918 | 0.8913 |
| libsvm | 0.9255 | 0.8753 | 0.8901 | 0.8971 | 0.8822 | 0.8896 |
| High recall | 0.6732 | 0.9562 | 0.7901 | 0.7967 | 0.9314 | 0.8588 |
| Higher recall | 0.5922 | 0.9622 | 0.7331 | 0.5202 | 0.9762 | 0.6787 |
| System | Evaluation sets | Test set | ||||
|---|---|---|---|---|---|---|
| Precision | Recall | F-score | Precision | Recall | F-score | |
| Logistic | 0.8867 | 0.8979 | 0.8923 | 0.8752 | 0.9129 | 0.8937 |
| Huber SVM | 0.9091 | 0.8626 | 0.8853 | 0.8908 | 0.8918 | 0.8913 |
| libsvm | 0.9255 | 0.8753 | 0.8901 | 0.8971 | 0.8822 | 0.8896 |
| High recall | 0.6732 | 0.9562 | 0.7901 | 0.7967 | 0.9314 | 0.8588 |
| Higher recall | 0.5922 | 0.9622 | 0.7331 | 0.5202 | 0.9762 | 0.6787 |
The internal evaluation values are averaged between the two evaluation sets. The highest value is shown in bold.
Results for our ensemble systems on the CEMP task as measured by precision (P), recall (R) and f-score (F)
| System | Evaluation sets | Test set | ||||
|---|---|---|---|---|---|---|
| Precision | Recall | F-score | Precision | Recall | F-score | |
| Logistic | 0.8867 | 0.8979 | 0.8923 | 0.8752 | 0.9129 | 0.8937 |
| Huber SVM | 0.9091 | 0.8626 | 0.8853 | 0.8908 | 0.8918 | 0.8913 |
| libsvm | 0.9255 | 0.8753 | 0.8901 | 0.8971 | 0.8822 | 0.8896 |
| High recall | 0.6732 | 0.9562 | 0.7901 | 0.7967 | 0.9314 | 0.8588 |
| Higher recall | 0.5922 | 0.9622 | 0.7331 | 0.5202 | 0.9762 | 0.6787 |
| System | Evaluation sets | Test set | ||||
|---|---|---|---|---|---|---|
| Precision | Recall | F-score | Precision | Recall | F-score | |
| Logistic | 0.8867 | 0.8979 | 0.8923 | 0.8752 | 0.9129 | 0.8937 |
| Huber SVM | 0.9091 | 0.8626 | 0.8853 | 0.8908 | 0.8918 | 0.8913 |
| libsvm | 0.9255 | 0.8753 | 0.8901 | 0.8971 | 0.8822 | 0.8896 |
| High recall | 0.6732 | 0.9562 | 0.7901 | 0.7967 | 0.9314 | 0.8588 |
| Higher recall | 0.5922 | 0.9622 | 0.7331 | 0.5202 | 0.9762 | 0.6787 |
The internal evaluation values are averaged between the two evaluation sets. The highest value is shown in bold.
GPRO task results
The GPRO task evaluation was also in terms of precision, recall and f-score, again requiring the exact span to be considered a true positive. However, only mentions that can be mapped to an identifier (type 1) are evaluated. Recognizing the mentions that cannot be mapped to a specific identifier (type 2) is therefore highly important, but we unfortunately found these mentions to be highly ambiguous with the type 1 mentions. We found that the CRF model could not differentiate well between the two types (models 2 and 3), but combining the types and refining the result in post-processing provided better performance (model 1). Adding the recognition result of GNormPlus as an additional feature in the CRF models increased recall about 4–6%, but reduced precision significantly. We produced two versions that aggregated the recognition results with a majority voting strategy. The last row in Table 5 aggregated the results of all five models, and obtained highest f-score (0.8137). All models were internally evaluated using the development data. The models were then retrained to include the development data for application to the test set.
Micro-averaged results for each model on the GPRO task test set, as measured by precision (P), recall (R) and f-score (F).
| Methods | Precision | Recall | F-score |
|---|---|---|---|
| M1 | 0.7835 | 0.8302 | 0.8062 |
| M2 | 0.8224 | 0.7852 | 0.8034 |
| M4 | 0.7677 | 0.8502 | 0.8069 |
| Majority voting based on M1–M4 | 0.8059 | 0.7982 | 0.8020 |
| Majority voting based on M1–M5 | 0.8143 | 0.8141 | 0.8137 |
| Methods | Precision | Recall | F-score |
|---|---|---|---|
| M1 | 0.7835 | 0.8302 | 0.8062 |
| M2 | 0.8224 | 0.7852 | 0.8034 |
| M4 | 0.7677 | 0.8502 | 0.8069 |
| Majority voting based on M1–M4 | 0.8059 | 0.7982 | 0.8020 |
| Majority voting based on M1–M5 | 0.8143 | 0.8141 | 0.8137 |
The highest value is shown in bold.
Micro-averaged results for each model on the GPRO task test set, as measured by precision (P), recall (R) and f-score (F).
| Methods | Precision | Recall | F-score |
|---|---|---|---|
| M1 | 0.7835 | 0.8302 | 0.8062 |
| M2 | 0.8224 | 0.7852 | 0.8034 |
| M4 | 0.7677 | 0.8502 | 0.8069 |
| Majority voting based on M1–M4 | 0.8059 | 0.7982 | 0.8020 |
| Majority voting based on M1–M5 | 0.8143 | 0.8141 | 0.8137 |
| Methods | Precision | Recall | F-score |
|---|---|---|---|
| M1 | 0.7835 | 0.8302 | 0.8062 |
| M2 | 0.8224 | 0.7852 | 0.8034 |
| M4 | 0.7677 | 0.8502 | 0.8069 |
| Majority voting based on M1–M4 | 0.8059 | 0.7982 | 0.8020 |
| Majority voting based on M1–M5 | 0.8143 | 0.8141 | 0.8137 |
The highest value is shown in bold.
CEMP task discussion
The strong performance obtained by the ensemble of chemical NER systems on the CEMP task is notable, given the perceived importance of the diversity of the models. Despite the broadly similar approach, however, the systems do exhibit many differences. These differences include varying strategies for sentence splitting, tokenization and also the toolkit used to implement conditional random fields (CRF). The feature sets used are also broadly similar, though becas[chemical] includes an extensive dictionary, and both the Wuhan tagger and BANNER-CHEMDNER include distributional semantics features. All systems use a token-level CRF model, but the Wuhan tagger combines this with a character level CRF model. All systems perform some level of abbreviation resolution and parenthesis post-processing, though both tmChem models and the Wuhan tagger attempt to revise the mention boundaries so the parenthesis will be balanced rather than always drop a mention with unbalanced parenthesis. These differences make it likely that any errors made by one model will frequently be different than the others and their strengths will complement each other.
While the performance gains obtained by combining independent systems into an ensemble are substantial, there are several disadvantages. Creating an ensemble requires a significant engineering effort: each system must be set up, configured, retrained, and adapted to the necessary data format(s). Each system then processes the data independently, requiring proportionally greater computing time. Finally, we found the results to be difficult to interpret: because ensembles work by ‘averaging’ out the errors of many individual systems, the remaining errors are obscured.
GPRO task discussion
Despite our best efforts, several types of errors remain. We manually analyzed the errors against the development set. We grouped our errors into several categories, as shown in Table 6 . The most common type of error appears to be due to boundary issues since the evaluation required exact match. Most boundary errors occur where a gene (e.g. ‘NPY1’) is nested within a larger gene mention (e.g. ‘NPY1 receptors’). Integrating gene/protein normalization may be able to address this issue: ‘NPY1’ and ‘NPY1 receptor’ are different genes, the prediction should therefore be the longer span (‘NPY1 receptor’).
The error analysis on 5-fold cross validation of the GPRO development set.
| Error type | Example | FPs | FNs | ||
|---|---|---|---|---|---|
| Incorrect boundary | ‘NPY1 receptors’ | 383 | (38.96%) | 405 | (51.92%) |
| Gene/family/domain confusion | ‘progesterone receptors’ | 188 | (19.13%) | 43 | (5.51%) |
| Not a gene mention | ‘MRSA’ | 226 | (22.99%) | ||
| Missed gene mention | ‘CB1’ | 175 | (22.44%) | ||
| Annotation inconsistency | ‘Alk1’ | 186 | (18.92%) | 157 | (20.13%) |
| Others | 1 | (0.10%) | 5 | (0.64%) | |
| Total | 983 | (100.00%) | 780 | (100.00%) |
| Error type | Example | FPs | FNs | ||
|---|---|---|---|---|---|
| Incorrect boundary | ‘NPY1 receptors’ | 383 | (38.96%) | 405 | (51.92%) |
| Gene/family/domain confusion | ‘progesterone receptors’ | 188 | (19.13%) | 43 | (5.51%) |
| Not a gene mention | ‘MRSA’ | 226 | (22.99%) | ||
| Missed gene mention | ‘CB1’ | 175 | (22.44%) | ||
| Annotation inconsistency | ‘Alk1’ | 186 | (18.92%) | 157 | (20.13%) |
| Others | 1 | (0.10%) | 5 | (0.64%) | |
| Total | 983 | (100.00%) | 780 | (100.00%) |
The error analysis on 5-fold cross validation of the GPRO development set.
| Error type | Example | FPs | FNs | ||
|---|---|---|---|---|---|
| Incorrect boundary | ‘NPY1 receptors’ | 383 | (38.96%) | 405 | (51.92%) |
| Gene/family/domain confusion | ‘progesterone receptors’ | 188 | (19.13%) | 43 | (5.51%) |
| Not a gene mention | ‘MRSA’ | 226 | (22.99%) | ||
| Missed gene mention | ‘CB1’ | 175 | (22.44%) | ||
| Annotation inconsistency | ‘Alk1’ | 186 | (18.92%) | 157 | (20.13%) |
| Others | 1 | (0.10%) | 5 | (0.64%) | |
| Total | 983 | (100.00%) | 780 | (100.00%) |
| Error type | Example | FPs | FNs | ||
|---|---|---|---|---|---|
| Incorrect boundary | ‘NPY1 receptors’ | 383 | (38.96%) | 405 | (51.92%) |
| Gene/family/domain confusion | ‘progesterone receptors’ | 188 | (19.13%) | 43 | (5.51%) |
| Not a gene mention | ‘MRSA’ | 226 | (22.99%) | ||
| Missed gene mention | ‘CB1’ | 175 | (22.44%) | ||
| Annotation inconsistency | ‘Alk1’ | 186 | (18.92%) | 157 | (20.13%) |
| Others | 1 | (0.10%) | 5 | (0.64%) | |
| Total | 983 | (100.00%) | 780 | (100.00%) |
Other errors include confusion with different entity types and annotation inconsistency. First, in our output gene/protein mentions were often confused and labeled erroneously as family names, many of which have a very similar appearance. For example, our system incorrectly identified ‘progesterone receptors’ as a family name because of the plural. Integrating gene/protein normalization may also help address this error: since there is no family with the name ‘progesterone receptors,’ the mention refers to a protein mention. Secondly, the same entity mention may be annotated differently in the same (e.g. only one of the three fup1 gene mentions in CN1654074A is annotated as a protein mention) or different documents (e.g. VEGF is annotated as a protein mention in US20110014197, but a family name in US20090074761). The remaining errors (category ‘Others’) include incorrect handling of composite mentions or abbreviations.
Conclusion
We identified chemical entities in patents (the CEMP task) using an ensemble of open source chemical NER systems, combined using a straightforward classification approach. The individual systems were trained either using the CEMP training data—which only contains patents, the BioCreative IV CHEMDNER data—which only contains PubMed abstracts, or both. Because the annotation guidelines for the CHEMDNER and CEMP corpora were highly similar, all individual models produced higher recall (with similar precision) when trained on the combination of the two corpora rather than only on patents. Our ensemble approach produced significantly higher performance than any of the constituent models. The models used in the ensemble contained small differences in approach, and also differed in the data used for training. We believe that combining these two types of differences in a large ensemble averaged out unimportant differences, producing strong performance. In future work we intend to create a software tool to simplify creating ensemble systems using an interoperable data format, such as BioC ( 28 ).
We addressed the gene product and related object (GPRO) task using a machine learning approach based on adapting GNormPlus, a conditional random field named entity recognizer. In this task, only mentions that can be mapped to a specific identifier (type 1) should be returned. These are highly ambiguous with the mentions that cannot be mapped to a specific identifier (type 2), and we found that the named entity recognizer could not differentiate between the two. Instead, we obtained better performance by combining the two types and refining the result in post-processing. We found that adding gene normalization as a feature increased recall, but results in a significant drop in precision. The highest performance we obtained was by aggregating the result of all models with a simple majority voting strategy. This task focuses only on gene/protein recognition; in future research we will focus on the gene/protein normalization to multiple resources (e.g., NCBI Gene and UniProt).
Acknowledgements
The authors thank the organizers of the BioCreative 5 CHEMDNER task.
Funding
This research is funded by the National Institutes of Health Intramural Research Program, National Library of Medicine (R.L., C.H., Z.L.). C.Z. was a summer intern at the NCBI/NIH and supported by an NIH Intramural Research Training Award.
Conflict of interest . None declared.
References
Author notes
These authors contributed equally to this work.
Citation details: Leaman,R., Wei,C.-H., Zou,C. et al. Mining chemical patents with an ensemble of open systems. Database (2016) Vol. 2016: article ID baw065; doi:10.1093/database/baw065

{kind=link}
{kind=link}